

Review
doi: 10.1186/s13059-017-1319-7.
Alignment-free sequence comparison: benefits, applications, and tools
Affiliations
- PMID: 28974235
- PMCID: PMC5627421
- DOI: 10.1186/s13059-017-1319-7
Item in Clipboard
Review
Alignment-free sequence comparison: benefits, applications, and tools
Genome Biol.
.
Abstract
Alignment-free sequence analyses have been applied to problems ranging from whole-genome phylogeny to the classification of protein families, identification of horizontally transferred genes, and detection of recombined sequences. The strength of these methods makes them particularly useful for next-generation sequencing data processing and analysis. However, many researchers are unclear about how these methods work, how they compare to alignment-based methods, and what their potential is for use for their research. We address these questions and provide a guide to the currently available alignment-free sequence analysis tools.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Figures
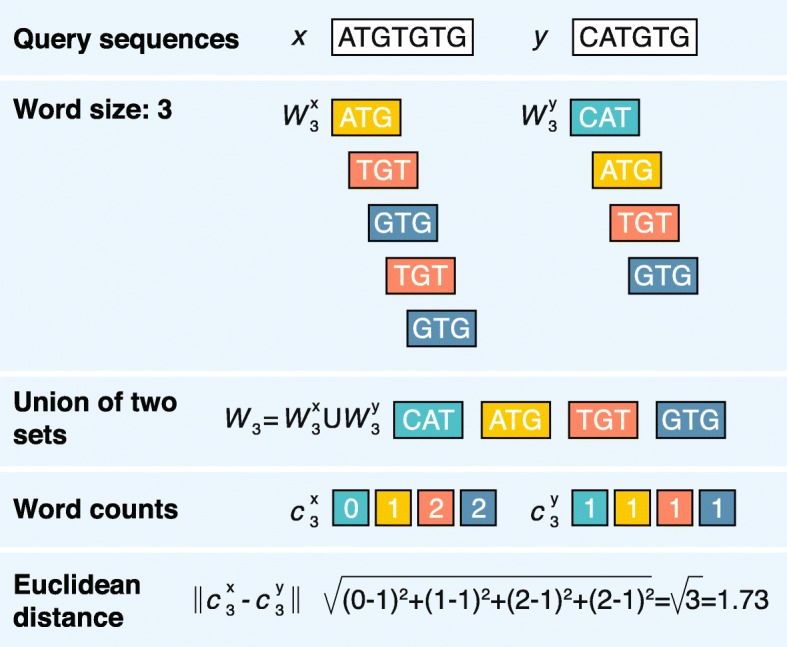
Alignment-free calculation of the word-based distance between two sample DNA sequences ATGTGTG and CATGTG using the Euclidean distance
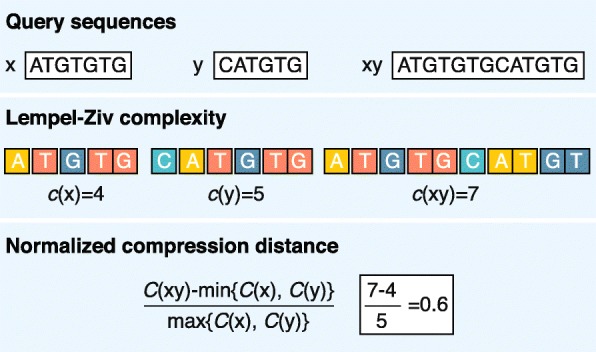
Alignment-free calculation of the normalized compression distance using the Lempel–Ziv complexity estimation algorithm. Lempel–Ziv complexity counts the number of different words in sequence when scanned from left to right (e.g., for s = ATGTGTG, Lempel–Ziv complexity is 4: A|T|G|TG). Description of compression algorithms in alignment-free analysis has been reviewed extensively [63]
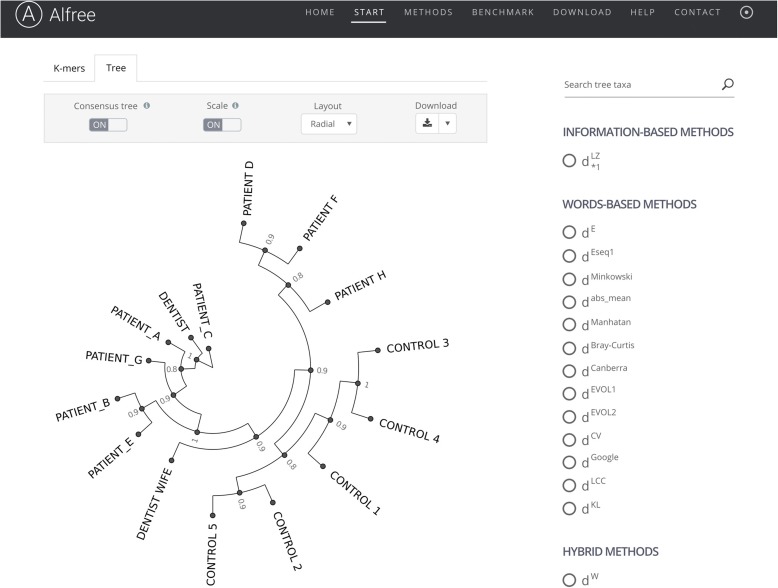
Snapshot of the results returned by the alignment-free web tool (Alfree) for “example 1”: HIV viral sequences obtained from dental patients in Florida [186]. Briefly, in the late 1980s some patients of an HIV-positive dentist in Florida were diagnosed as infected with HIV. An investigation by the Centers for Disease Control and Prevention did not uncover any hygiene lapses that could result in infection of patients. However, sequence comparison of the gene encoding gpg120 isolated from HIV strains from the dentist, his patients, and other individuals revealed that PATIENT_A, PATIENT_B, PATIENT_C, PATIENT_E, and PATIENT_G became infected while receiving dental care [183]. The phylogeny shown is based on the gp120 viral protein sequences from the dentist, the dentist’s wife (DENTIST WIFE), eight patients (PATIENT_A to PATIENT H), and five individuals that never had contact with the accused (CONTROL 1, 2, 3, 4, and 5). The sphylogram was obtained as a majority-rule consensus tree that summarizes the agreement across 15 alignment-free methods (support values in scale from 0 to 1 are shown for every node of the tree). The web interface of the Alfree portal also provides an example case of phylogenetic reconstruction of mitochondrial genomes of 12 primates. Several additional options are available to explore and visualize the sequence comparison results, including selection of individual method, re-rooting trees, changing tree layouts, as well as collapsing or expanding different parts of the tree
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
