

Mutations in Membrin/GOSR2 Reveal Stringent Secretory Pathway Demands of Dendritic Growth and Synaptic Integrity
- PMID: 28978487
- PMCID: PMC5640804
- DOI: 10.1016/j.celrep.2017.09.004
Mutations in Membrin/GOSR2 Reveal Stringent Secretory Pathway Demands of Dendritic Growth and Synaptic Integrity
Abstract
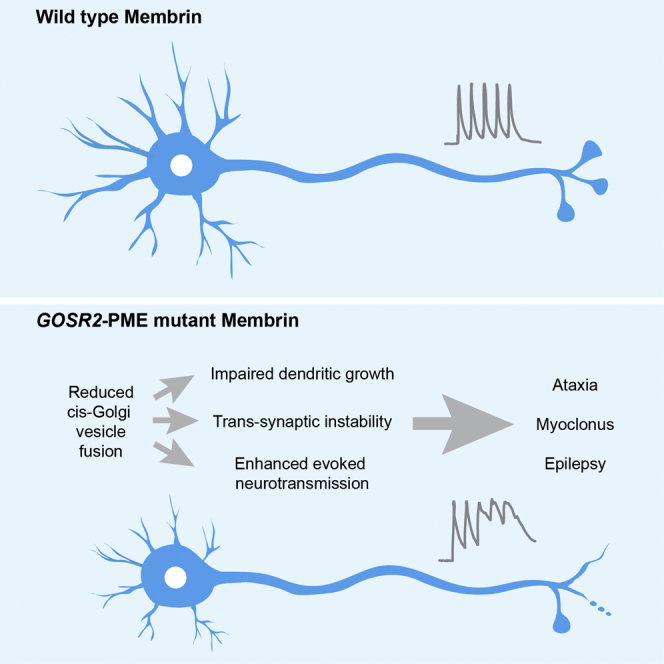
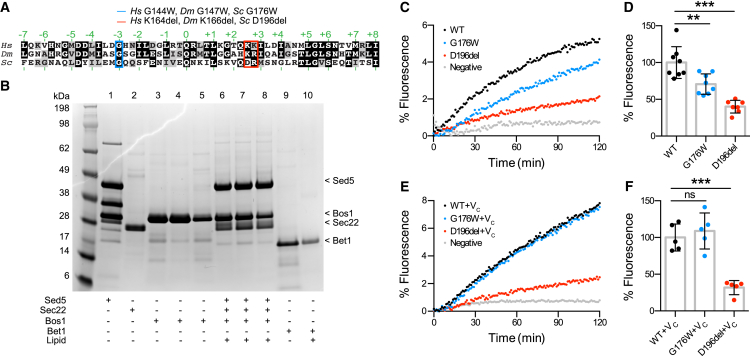
Mutations in the Golgi SNARE (SNAP [soluble NSF attachment protein] receptor) protein Membrin (encoded by the GOSR2 gene) cause progressive myoclonus epilepsy (PME). Membrin is a ubiquitous and essential protein mediating ER-to-Golgi membrane fusion. Thus, it is unclear how mutations in Membrin result in a disorder restricted to the nervous system. Here, we use a multi-layered strategy to elucidate the consequences of Membrin mutations from protein to neuron. We show that the pathogenic mutations cause partial reductions in SNARE-mediated membrane fusion. Importantly, these alterations were sufficient to profoundly impair dendritic growth in Drosophila models of GOSR2-PME. Furthermore, we show that Membrin mutations cause fragmentation of the presynaptic cytoskeleton coupled with transsynaptic instability and hyperactive neurotransmission. Our study highlights how dendritic growth is vulnerable even to subtle secretory pathway deficits, uncovers a role for Membrin in synaptic function, and provides a comprehensive explanatory basis for genotype-phenotype relationships in GOSR2-PME.
Keywords: GOSR2; GS27; Membrin; dendrite growth; progressive myoclonus epilepsy; synaptic integrity.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures






References
-
- Bishop D.L., Misgeld T., Walsh M.K., Gan W.-B., Lichtman J.W. Axon branch removal at developing synapses by axosome shedding. Neuron. 2004;44:651–661. - PubMed
-
- Boissé Lomax L., Bayly M.A., Hjalgrim H., Møller R.S., Vlaar A.M., Aaberg K.M., Marquardt I., Gandolfo L.C., Willemsen M., Kamsteeg E.-J. ‘North Sea’ progressive myoclonus epilepsy: phenotype of subjects with GOSR2 mutation. Brain. 2013;136:1146–1154. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
