

International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors
- PMID: 28978633
- PMCID: PMC5629631
- DOI: 10.1124/pr.117.014050
International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors
Abstract
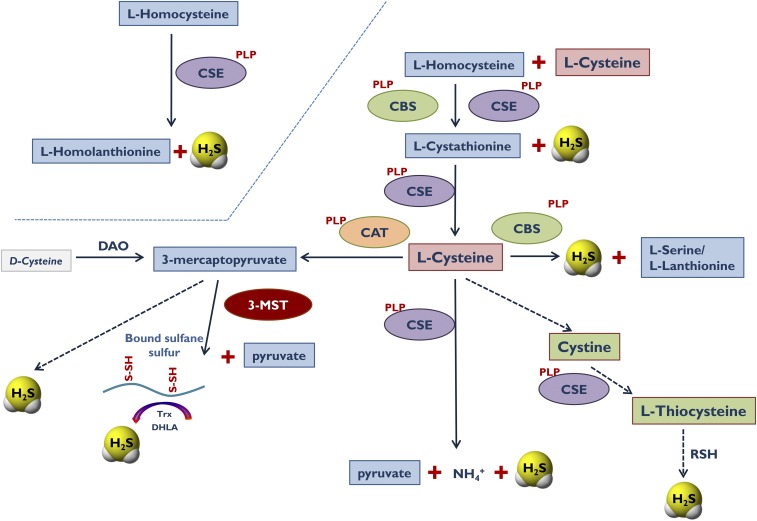
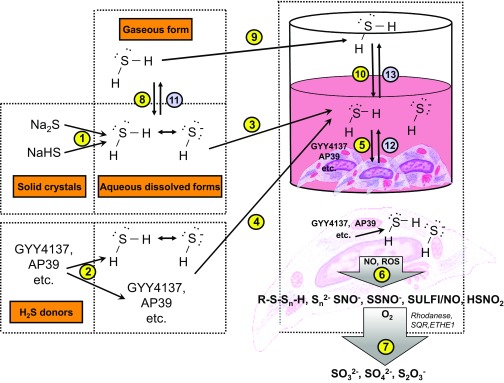
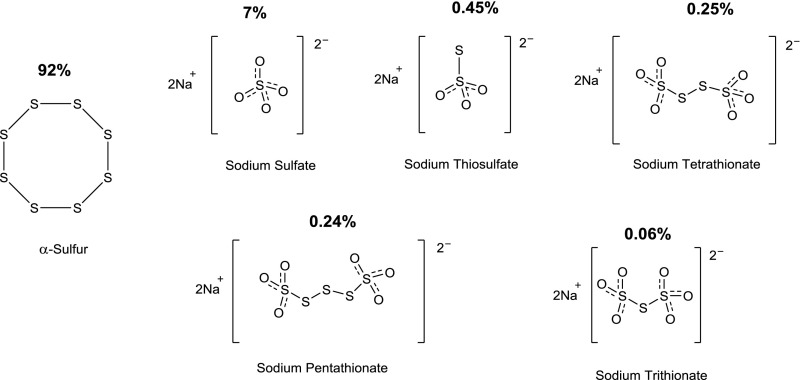
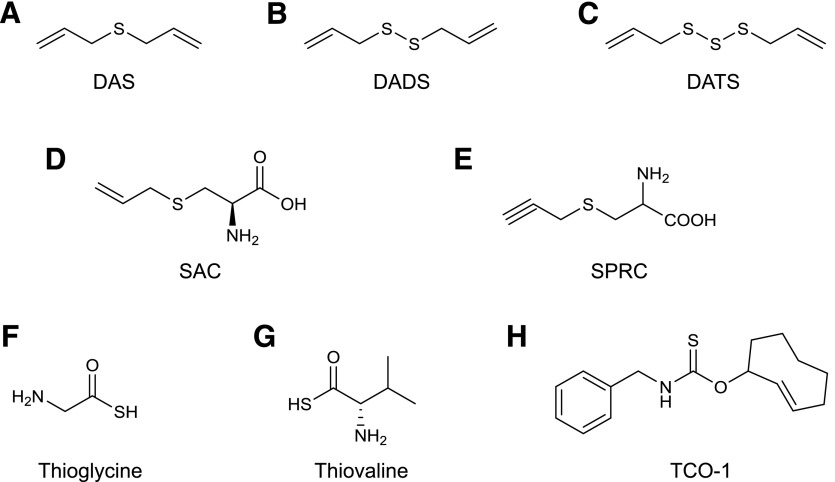
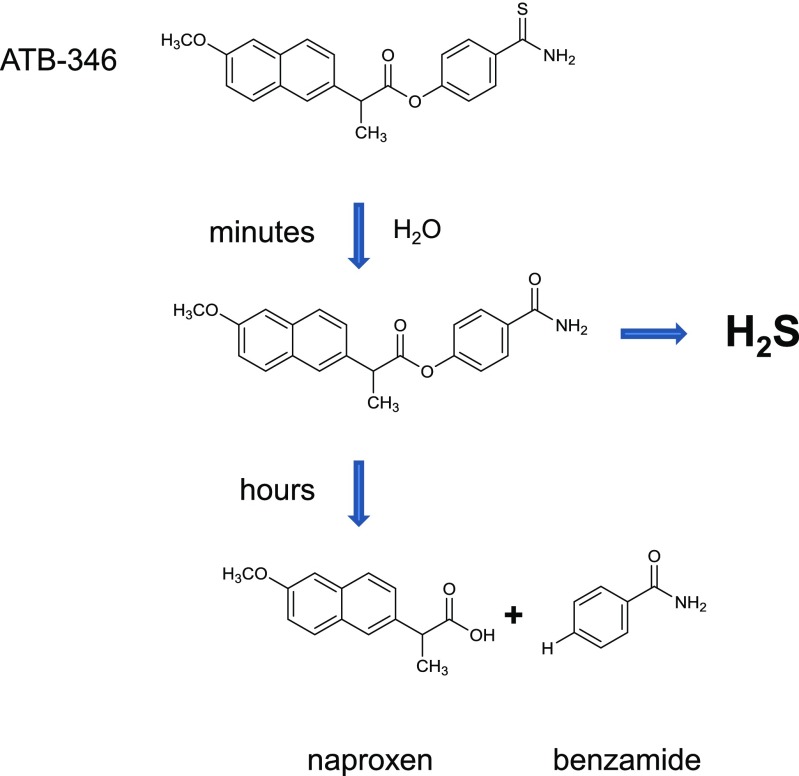
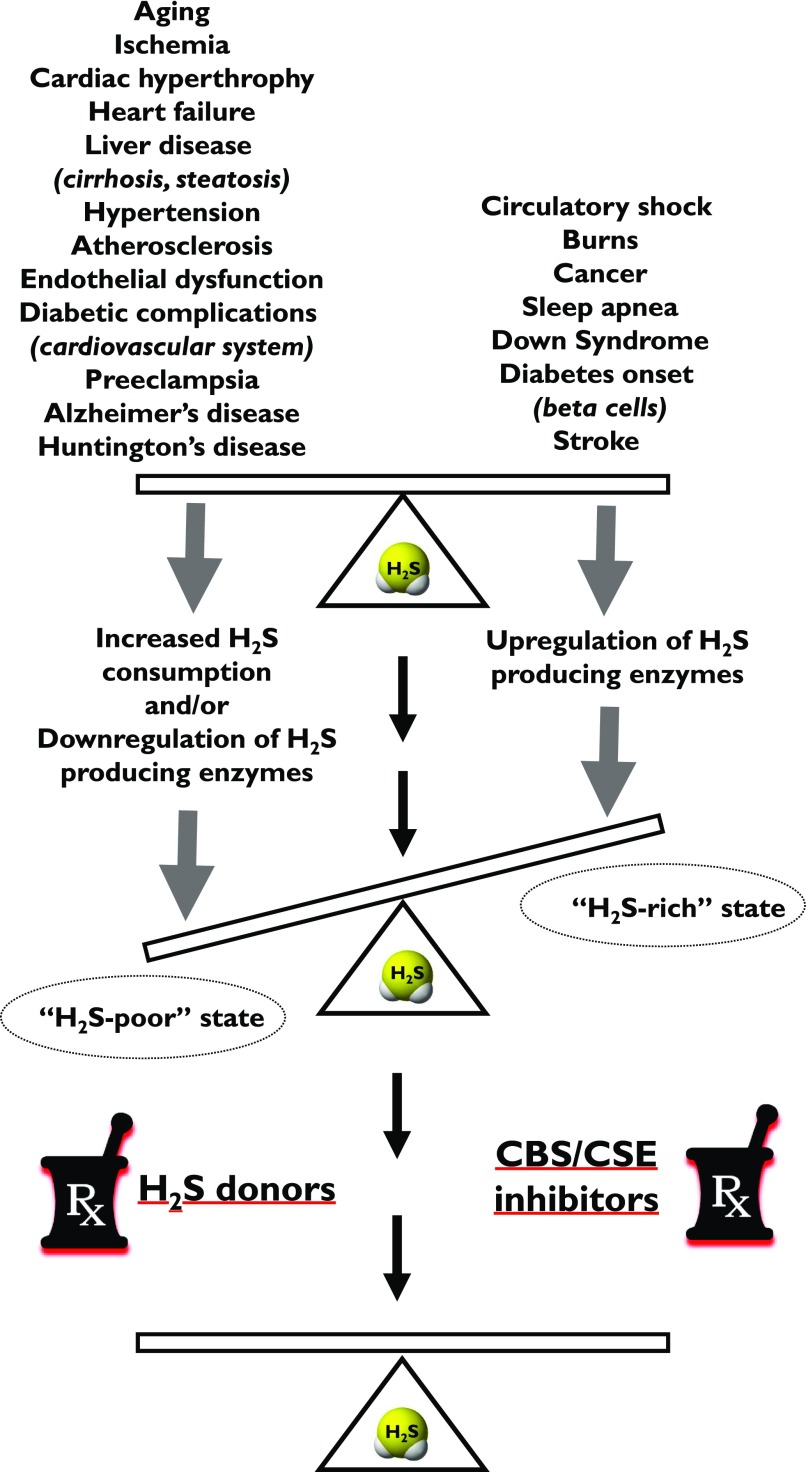
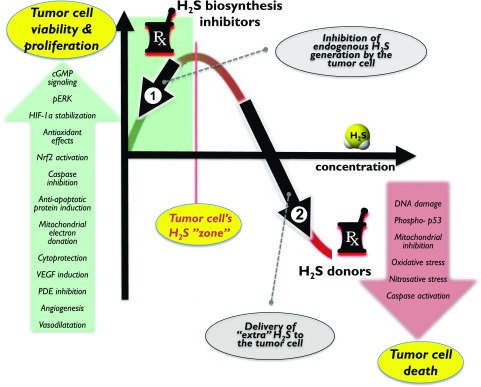
Over the last decade, hydrogen sulfide (H2S) has emerged as an important endogenous gasotransmitter in mammalian cells and tissues. Similar to the previously characterized gasotransmitters nitric oxide and carbon monoxide, H2S is produced by various enzymatic reactions and regulates a host of physiologic and pathophysiological processes in various cells and tissues. H2S levels are decreased in a number of conditions (e.g., diabetes mellitus, ischemia, and aging) and are increased in other states (e.g., inflammation, critical illness, and cancer). Over the last decades, multiple approaches have been identified for the therapeutic exploitation of H2S, either based on H2S donation or inhibition of H2S biosynthesis. H2S donation can be achieved through the inhalation of H2S gas and/or the parenteral or enteral administration of so-called fast-releasing H2S donors (salts of H2S such as NaHS and Na2S) or slow-releasing H2S donors (GYY4137 being the prototypical compound used in hundreds of studies in vitro and in vivo). Recent work also identifies various donors with regulated H2S release profiles, including oxidant-triggered donors, pH-dependent donors, esterase-activated donors, and organelle-targeted (e.g., mitochondrial) compounds. There are also approaches where existing, clinically approved drugs of various classes (e.g., nonsteroidal anti-inflammatories) are coupled with H2S-donating groups (the most advanced compound in clinical trials is ATB-346, an H2S-donating derivative of the non-steroidal anti-inflammatory compound naproxen). For pharmacological inhibition of H2S synthesis, there are now several small molecule compounds targeting each of the three H2S-producing enzymes cystathionine-β-synthase (CBS), cystathionine-γ-lyase, and 3-mercaptopyruvate sulfurtransferase. Although many of these compounds have their limitations (potency, selectivity), these molecules, especially in combination with genetic approaches, can be instrumental for the delineation of the biologic processes involving endogenous H2S production. Moreover, some of these compounds (e.g., cell-permeable prodrugs of the CBS inhibitor aminooxyacetate, or benserazide, a potentially repurposable CBS inhibitor) may serve as starting points for future clinical translation. The present article overviews the currently known H2S donors and H2S biosynthesis inhibitors, delineates their mode of action, and offers examples for their biologic effects and potential therapeutic utility.
Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics.
Figures
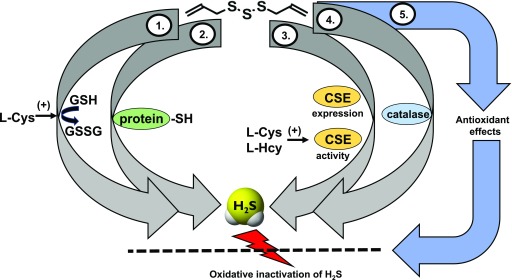
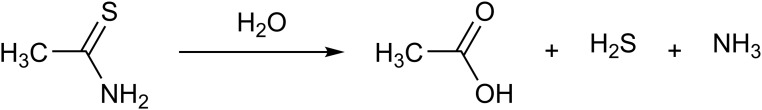
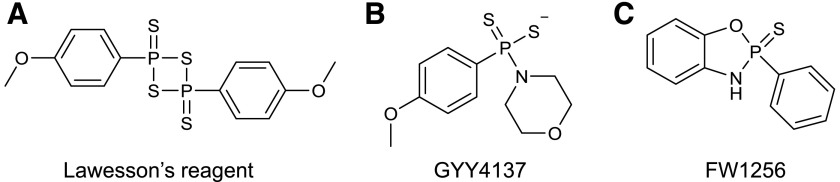
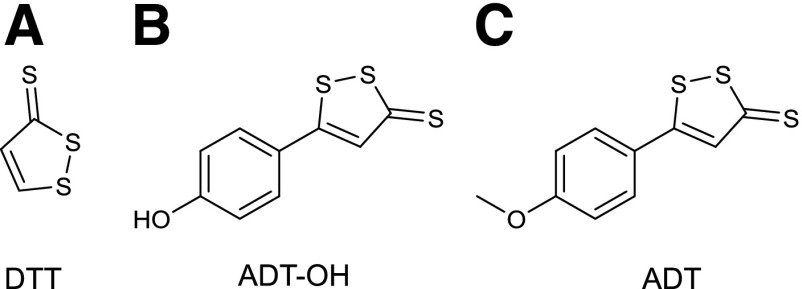
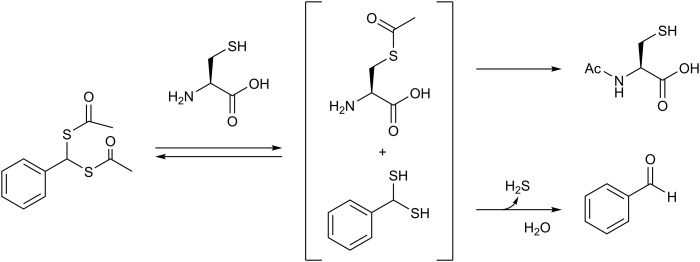
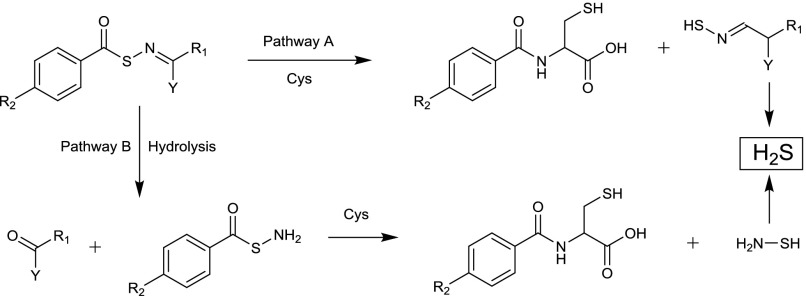
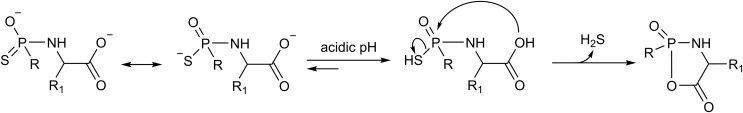
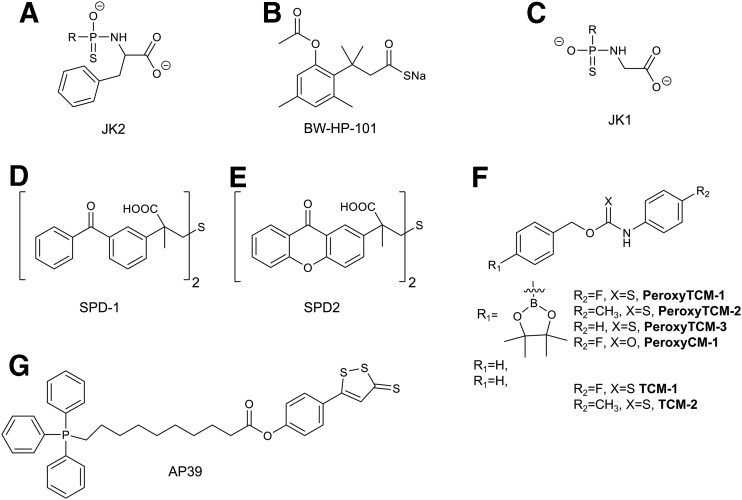
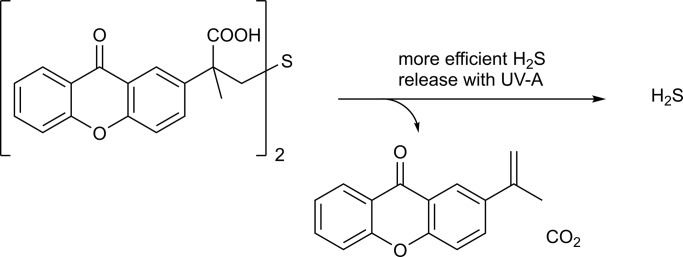
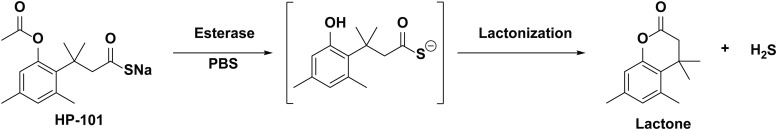
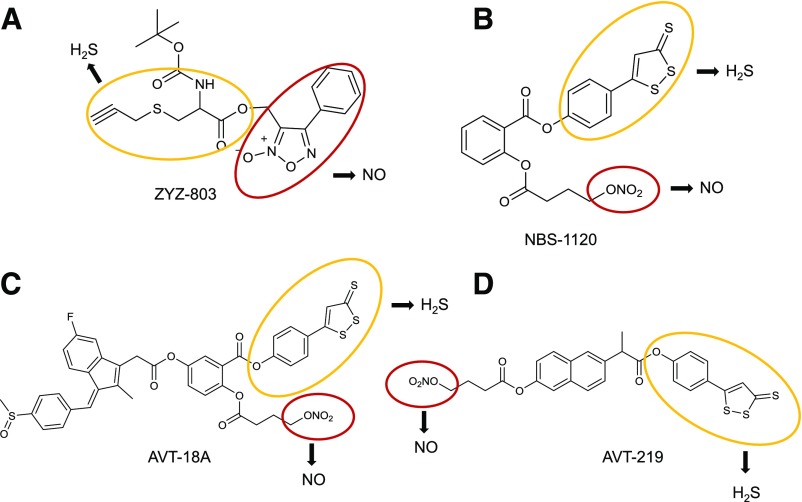
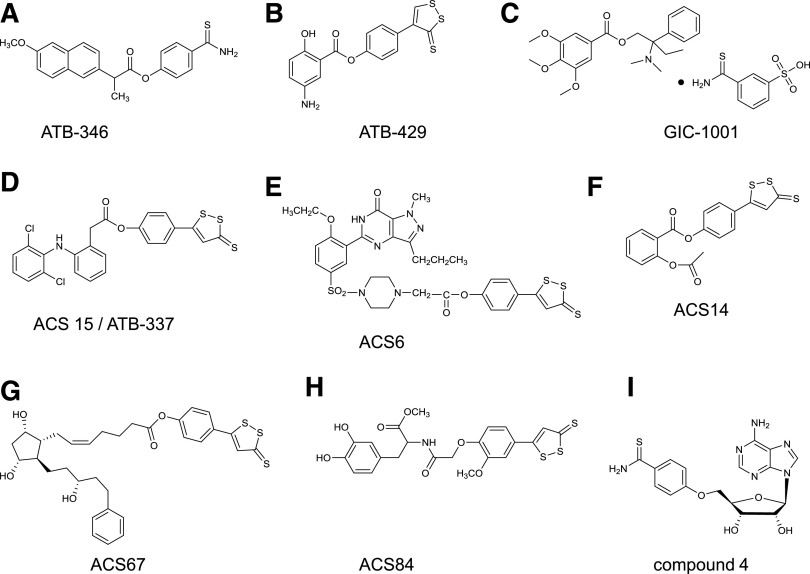
References
-
- Abdel-Salam E, Abdel-Meguid I, Korraa S. (2013) Assessment of immune function in Down syndrome patients. The Egyptian Journal of Medical Human Genetics 14:307–310.
-
- Abeles RH, Walsh CT. (1973) Acetylenic enzyme inactivators. Inactivation of gamma-cystathionase, in vitro and in vivo, by propargylglycine. J Am Chem Soc 95:6124–6125. - PubMed
-
- Abou-Hamdan A, Guedouari-Bounihi H, Lenoir V, Andriamihaja M, Blachier F, Bouillaud F. (2015) Oxidation of H2S in mammalian cells and mitochondria. Methods Enzymol 554:201–228. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
