

The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes
- PMID: 28978634
- PMCID: PMC5679174
- DOI: 10.1084/jem.20170689
The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes
Abstract
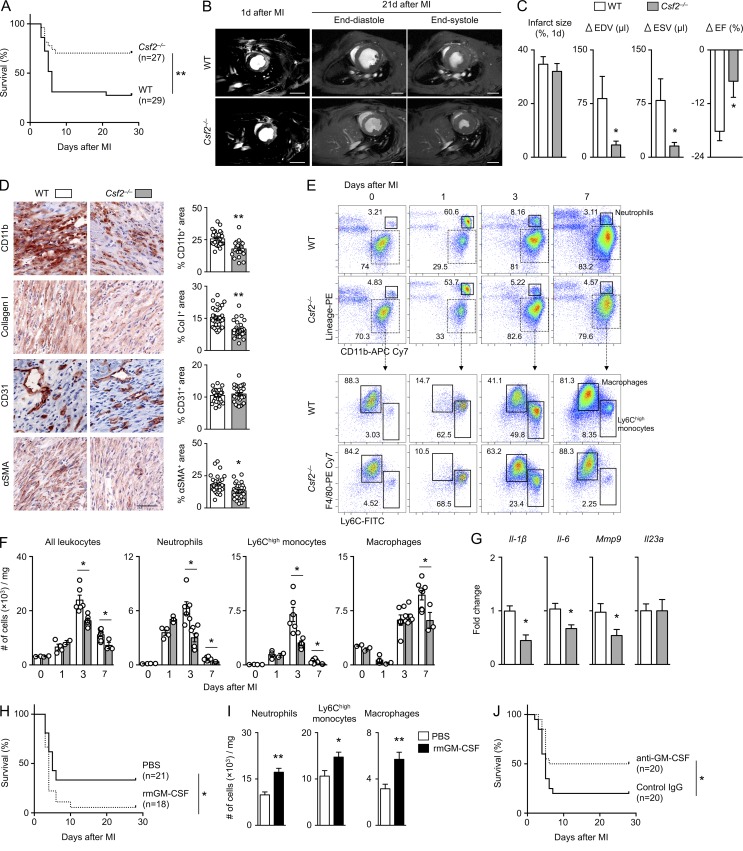
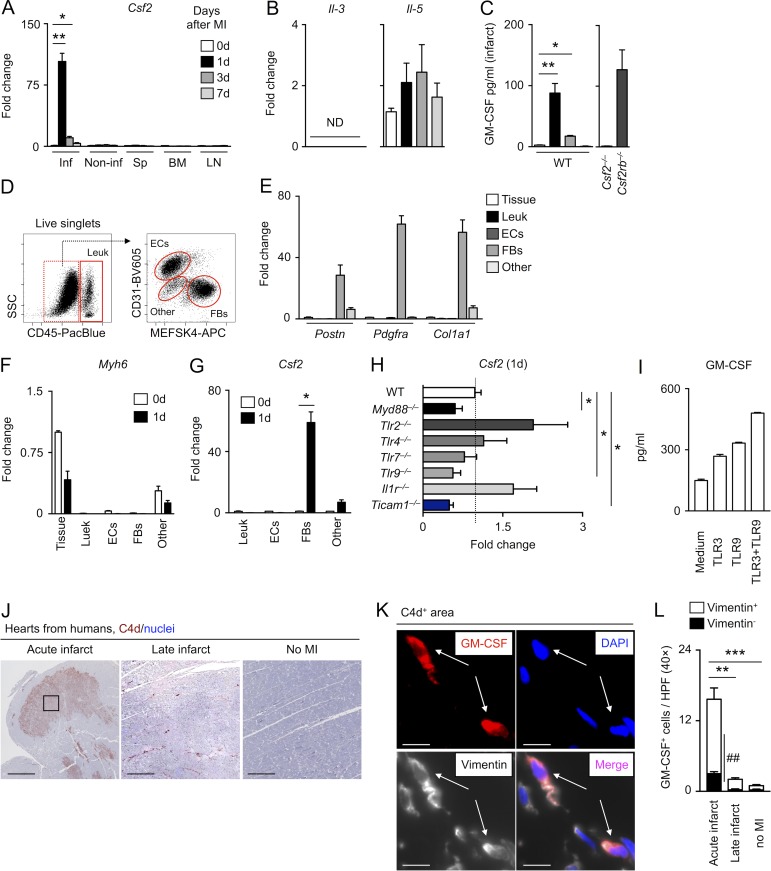
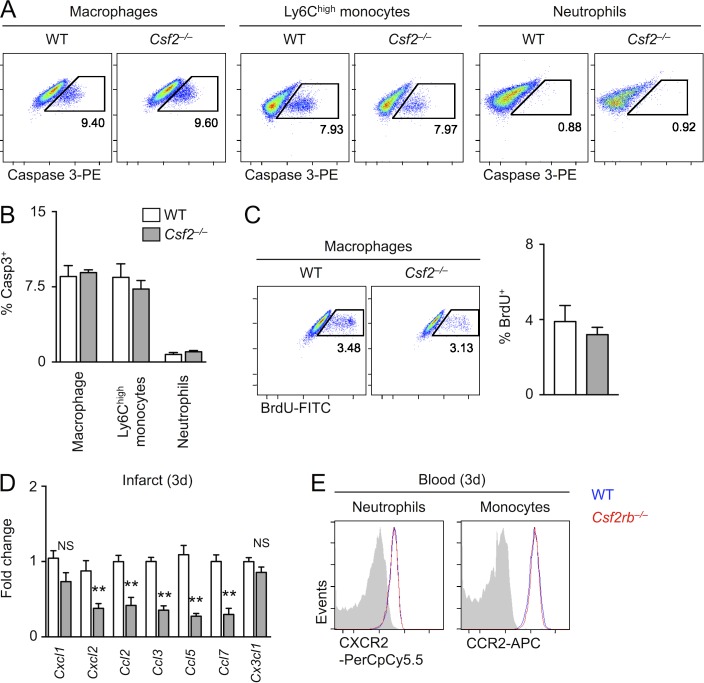
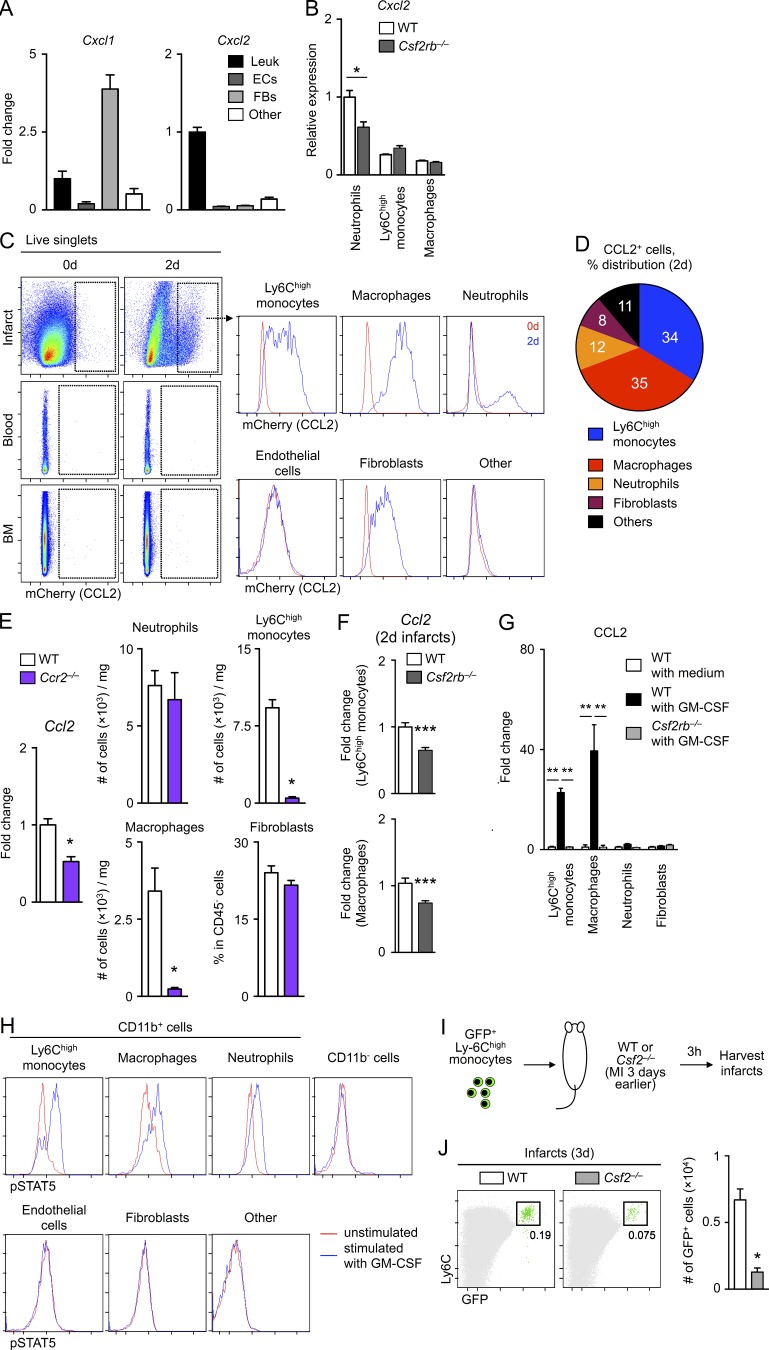
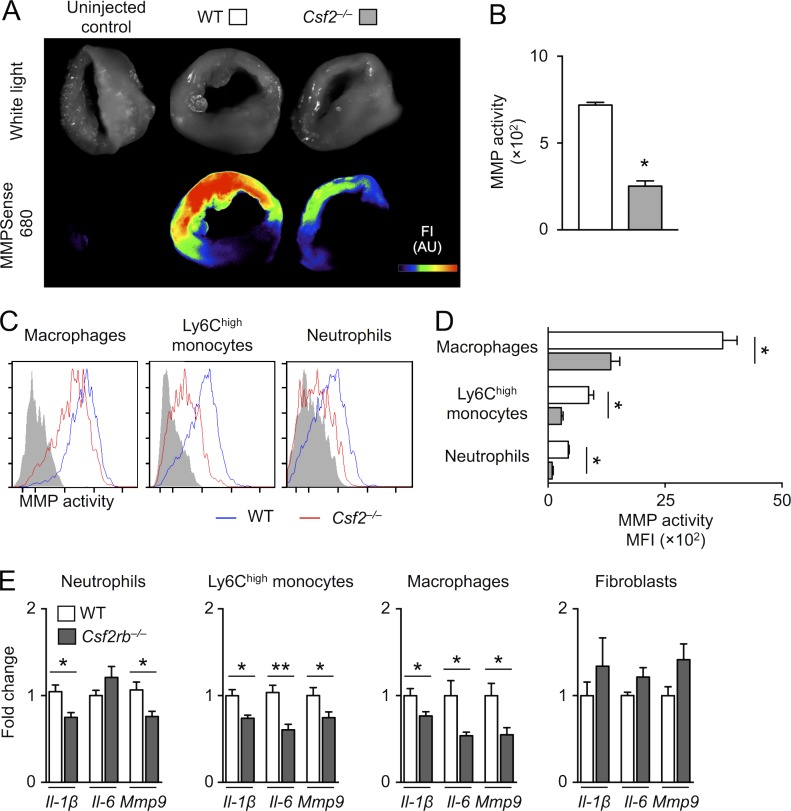
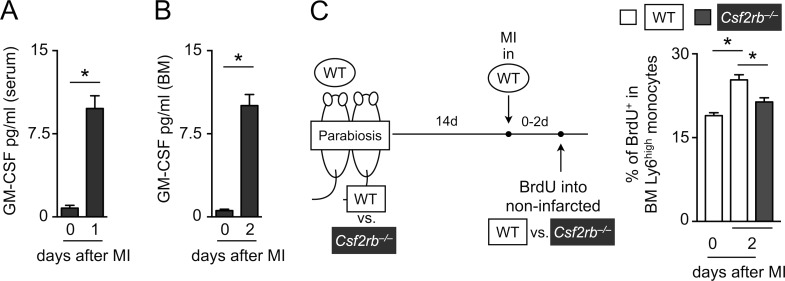
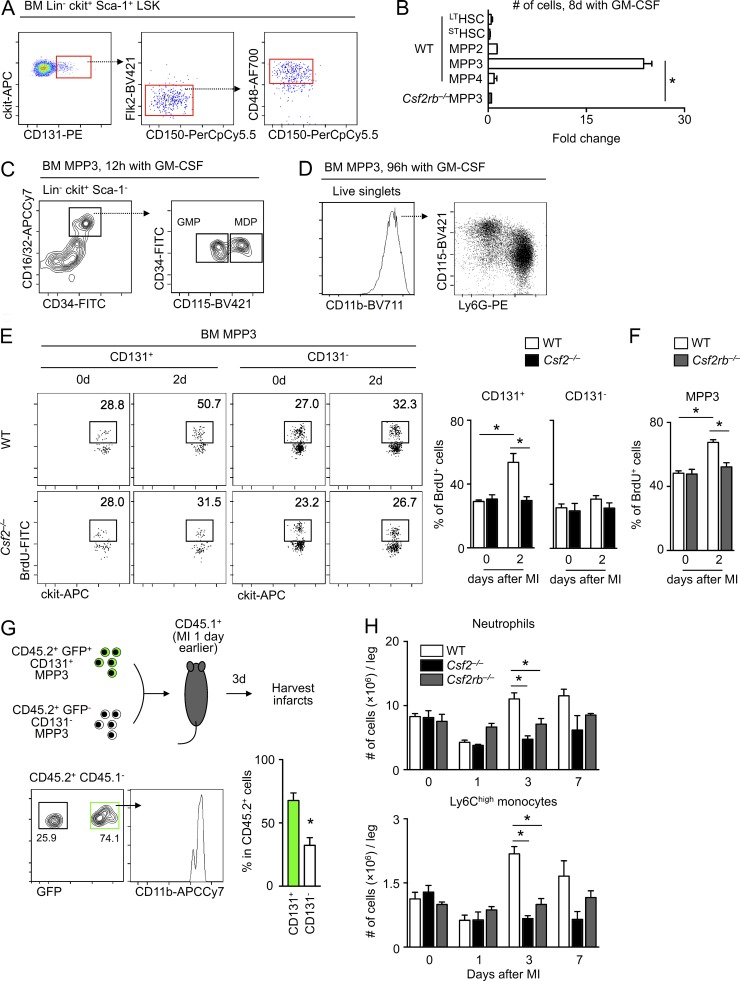
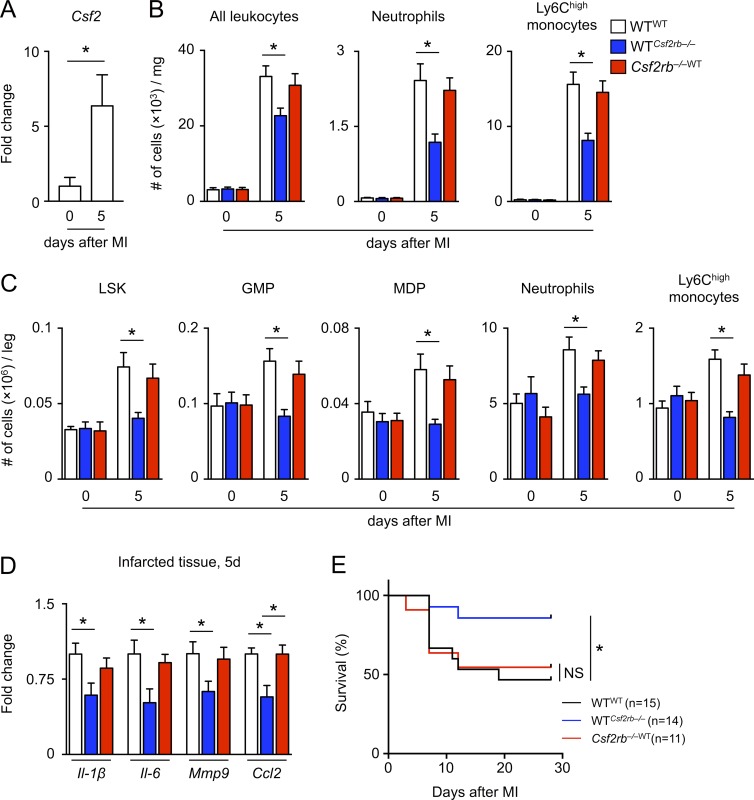
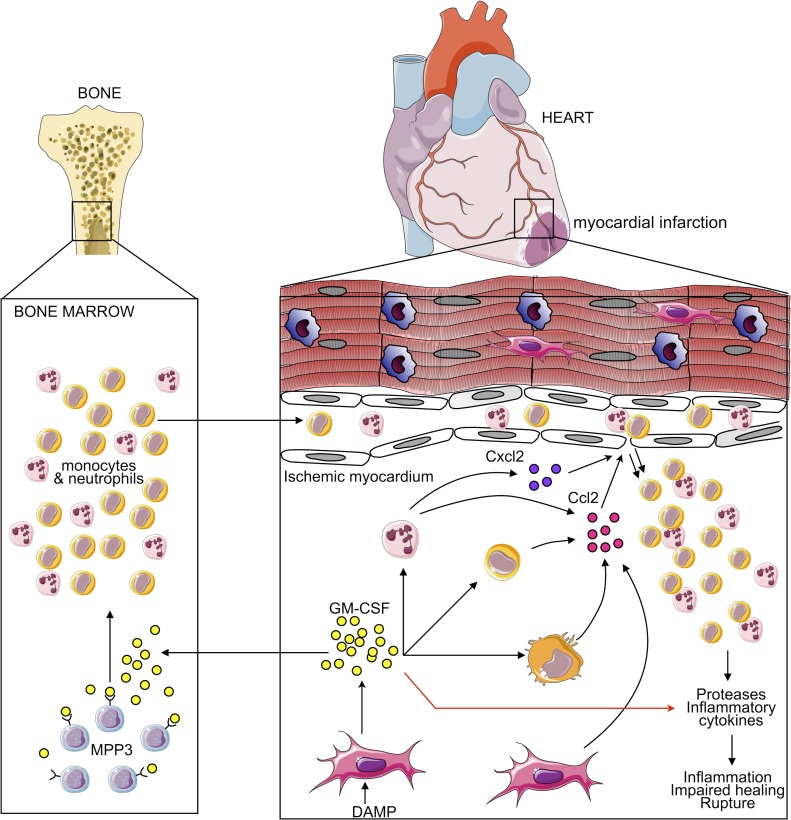
Myocardial infarction (MI) elicits massive inflammatory leukocyte recruitment to the heart. Here, we hypothesized that excessive leukocyte invasion leads to heart failure and death during acute myocardial ischemia. We found that shortly and transiently after onset of ischemia, human and mouse cardiac fibroblasts produce granulocyte/macrophage colony-stimulating factor (GM-CSF) that acts locally and distally to generate and recruit inflammatory and proteolytic cells. In the heart, fibroblast-derived GM-CSF alerts its neighboring myeloid cells to attract neutrophils and monocytes. The growth factor also reaches the bone marrow, where it stimulates a distinct myeloid-biased progenitor subset. Consequently, hearts of mice deficient in either GM-CSF or its receptor recruit fewer leukocytes and function relatively well, whereas mice producing GM-CSF can succumb from left ventricular rupture, a complication mitigated by anti-GM-CSF therapy. These results identify GM-CSF as both a key contributor to the pathogenesis of MI and a potential therapeutic target, bolstering the idea that GM-CSF is a major orchestrator of the leukocyte supply chain during inflammation.
© 2017 Anzai et al.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
