

NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data
- PMID: 28978689
- PMCID: PMC5679736
- DOI: 10.4049/jimmunol.1700893
NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data
Abstract
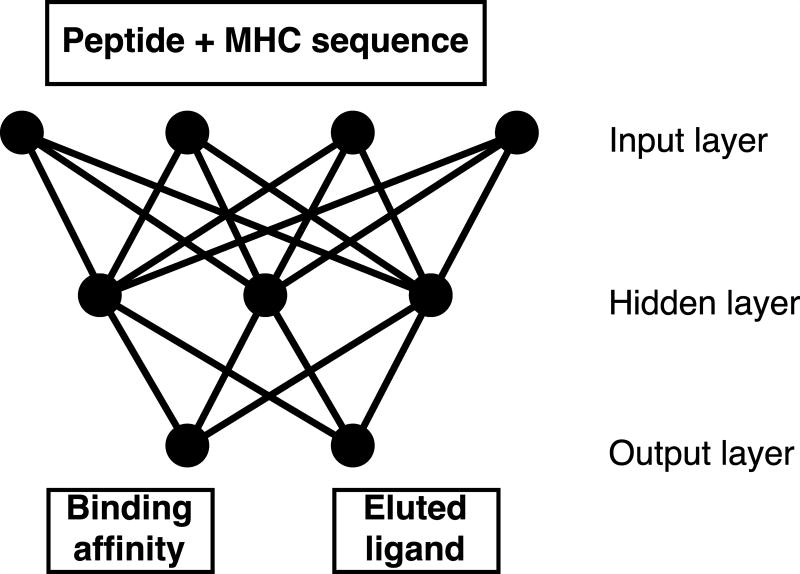
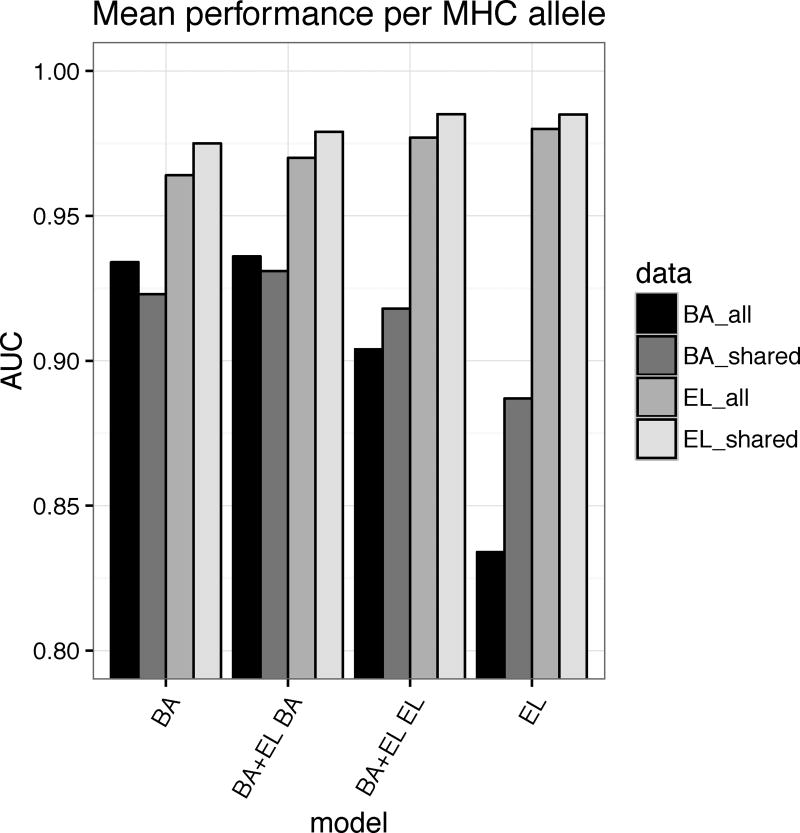
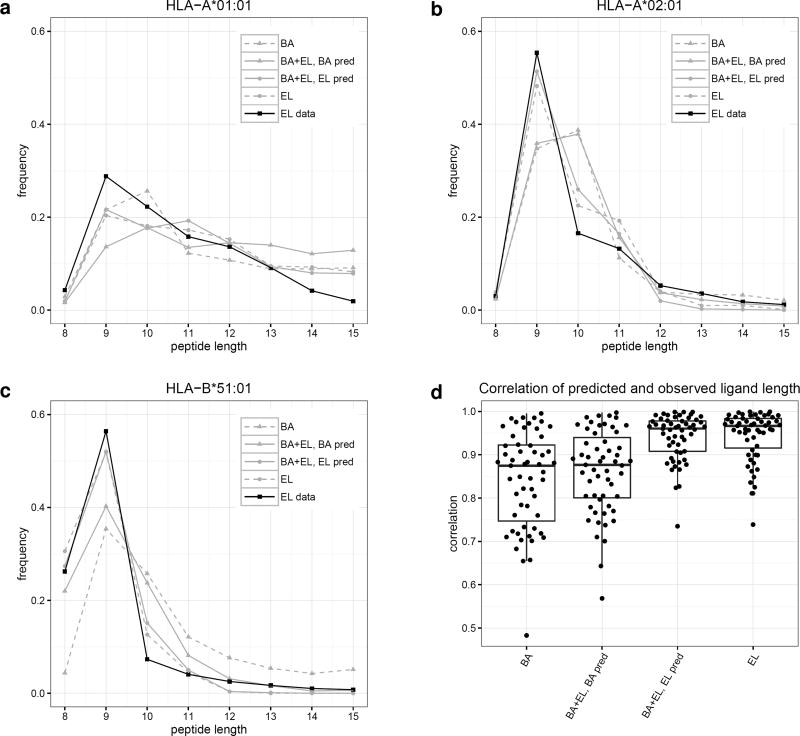
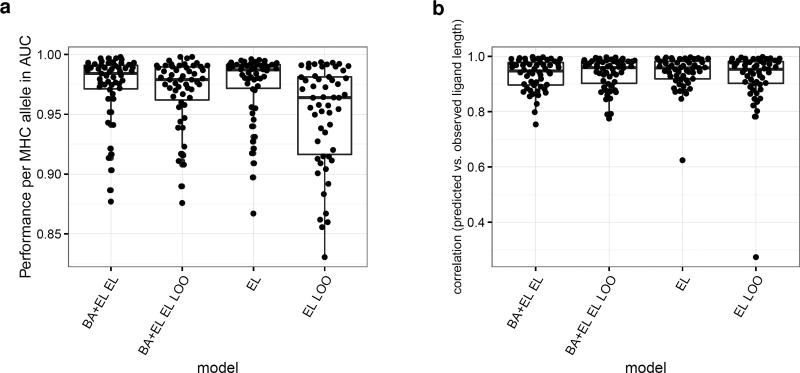
Cytotoxic T cells are of central importance in the immune system's response to disease. They recognize defective cells by binding to peptides presented on the cell surface by MHC class I molecules. Peptide binding to MHC molecules is the single most selective step in the Ag-presentation pathway. Therefore, in the quest for T cell epitopes, the prediction of peptide binding to MHC molecules has attracted widespread attention. In the past, predictors of peptide-MHC interactions have primarily been trained on binding affinity data. Recently, an increasing number of MHC-presented peptides identified by mass spectrometry have been reported containing information about peptide-processing steps in the presentation pathway and the length distribution of naturally presented peptides. In this article, we present NetMHCpan-4.0, a method trained on binding affinity and eluted ligand data leveraging the information from both data types. Large-scale benchmarking of the method demonstrates an increase in predictive performance compared with state-of-the-art methods when it comes to identification of naturally processed ligands, cancer neoantigens, and T cell epitopes.
Copyright © 2017 by The American Association of Immunologists, Inc.
Figures
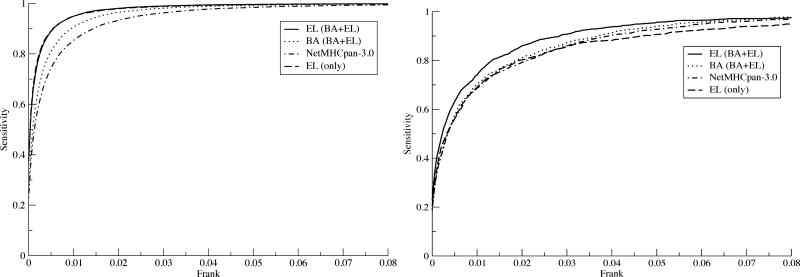
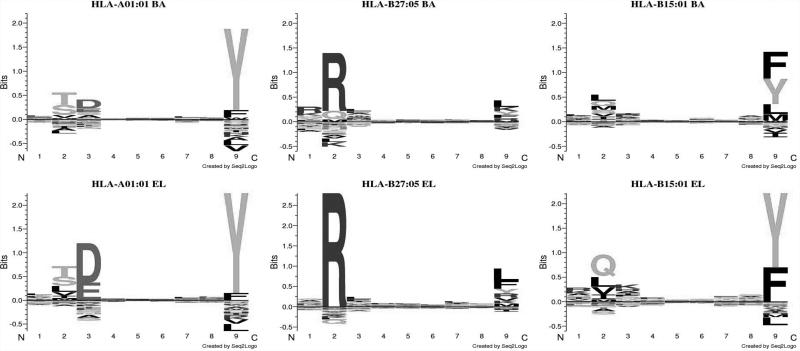
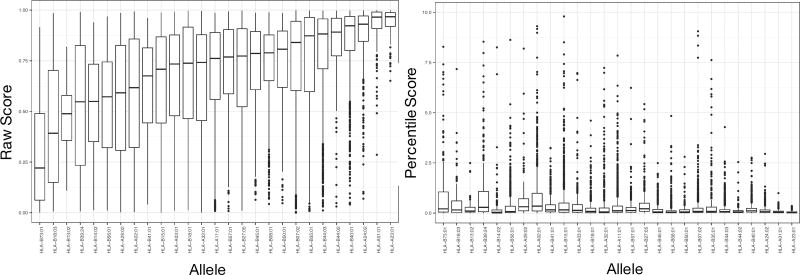
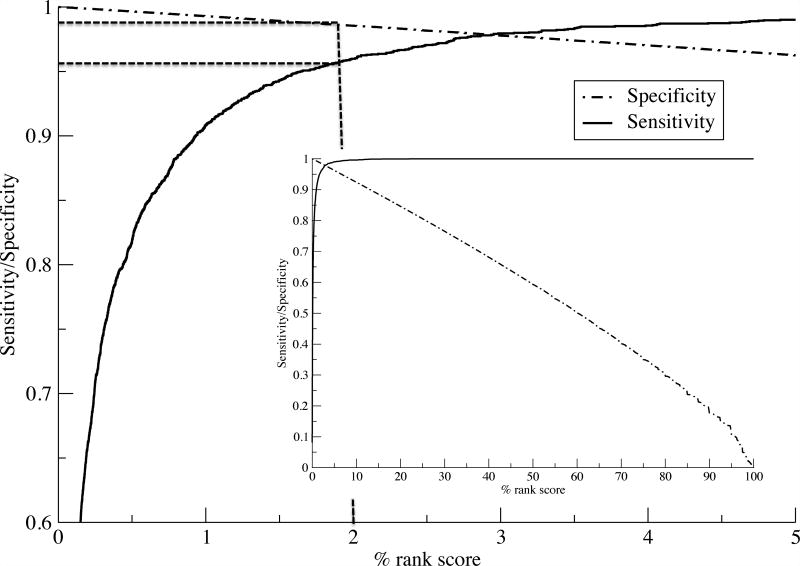
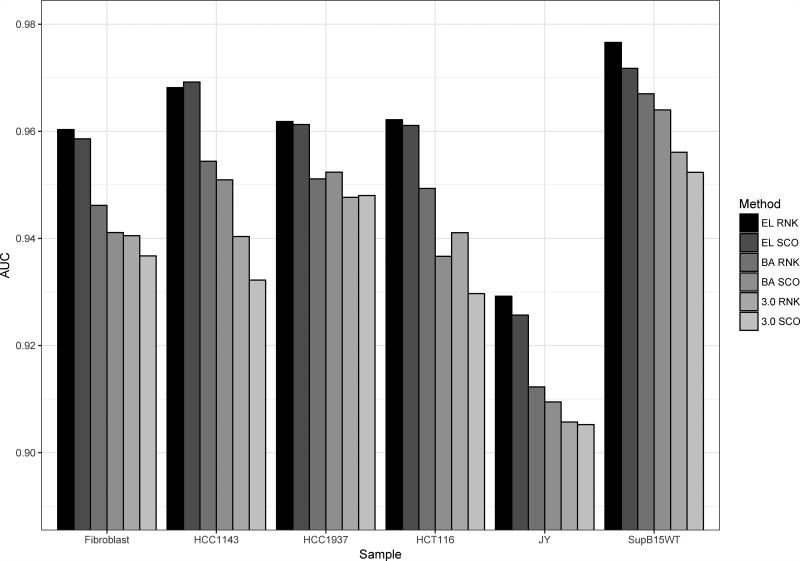
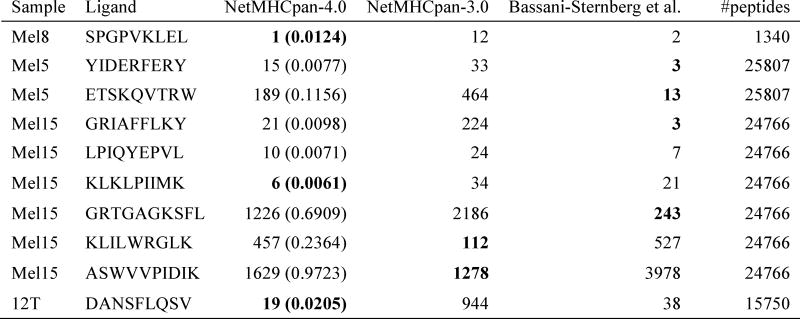
References
-
- Deres K, Schumacher TN, Wiesmuller KH, Stevanovic S, Greiner G, Jung G, Ploegh HL. Preferred size of peptides that bind to H-2 Kb is sequence dependent. Eur J Immunol. 1992;22:1603–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
