

Directing the use of DDR kinase inhibitors in cancer treatment
- PMID: 28984489
- PMCID: PMC6157710
- DOI: 10.1080/13543784.2017.1389895
Directing the use of DDR kinase inhibitors in cancer treatment
Abstract
Defects in the DNA damage response (DDR) drive the development of cancer by fostering DNA mutation but also provide cancer-specific vulnerabilities that can be exploited therapeutically. The recent approval of three different PARP inhibitors for the treatment of ovarian cancer provides the impetus for further developing targeted inhibitors of many of the kinases involved in the DDR, including inhibitors of ATR, ATM, CHEK1, CHEK2, DNAPK and WEE1. Areas covered: We summarise the current stage of development of these novel DDR kinase inhibitors, and describe which predictive biomarkers might be exploited to direct their clinical use. Expert opinion: Novel DDR inhibitors present promising candidates in cancer treatment and have the potential to elicit synthetic lethal effects. In order to fully exploit their potential and maximize their utility, identifying highly penetrant predictive biomarkers of single agent and combinatorial DDR inhibitor sensitivity are critical. Identifying the optimal drug combination regimens that could used with DDR inhibitors is also a key objective.
Keywords: Cancer; DNA damage response (DDR); cell cycle; kinase inhibitors; replication stress.
Figures
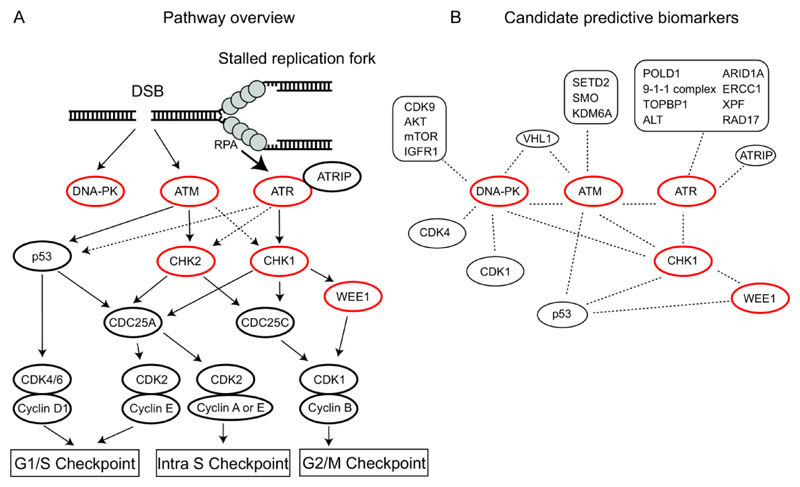
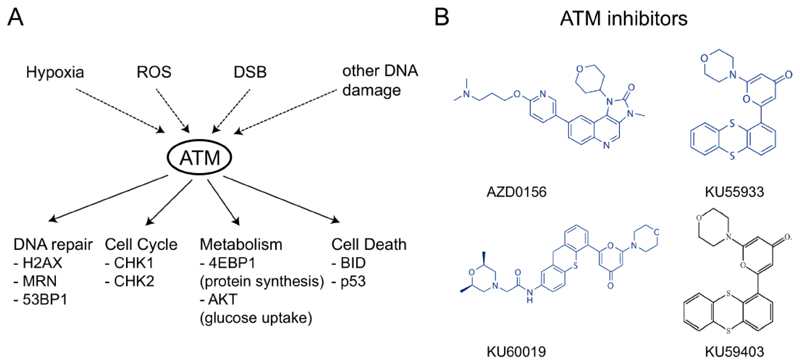
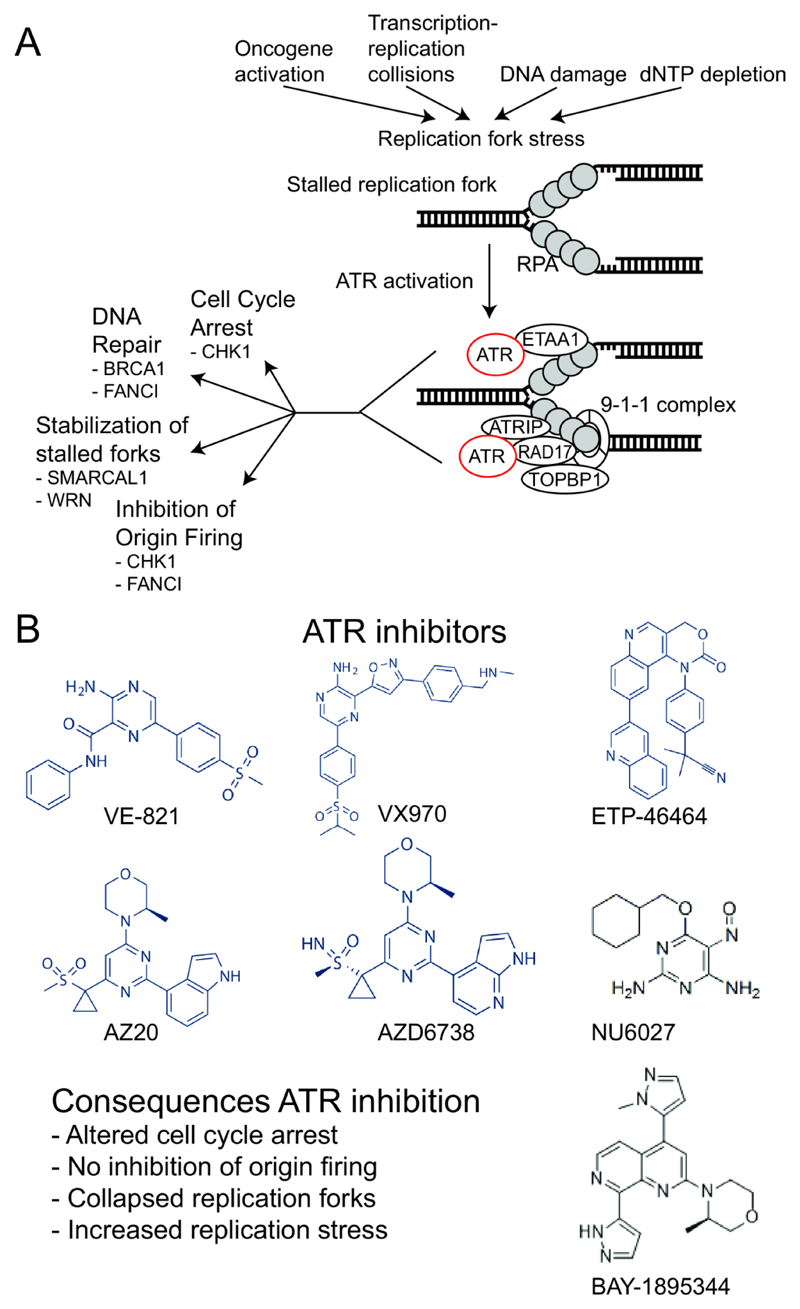
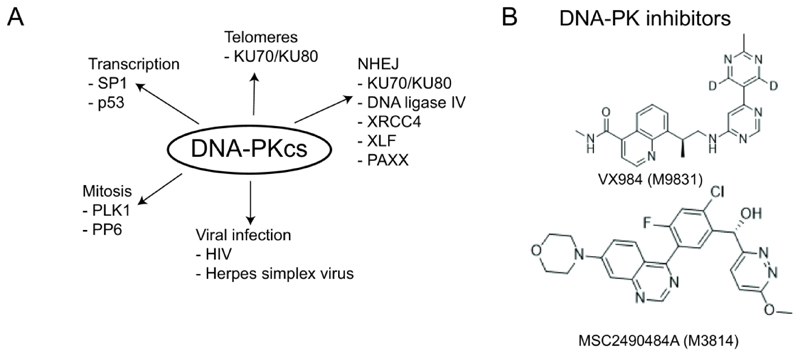
References
-
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. - PubMed
-
- Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. - PubMed
-
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous