

The Effects of Captivity on the Mammalian Gut Microbiome
- PMID: 28985326
- PMCID: PMC5978021
- DOI: 10.1093/icb/icx090
The Effects of Captivity on the Mammalian Gut Microbiome
Abstract
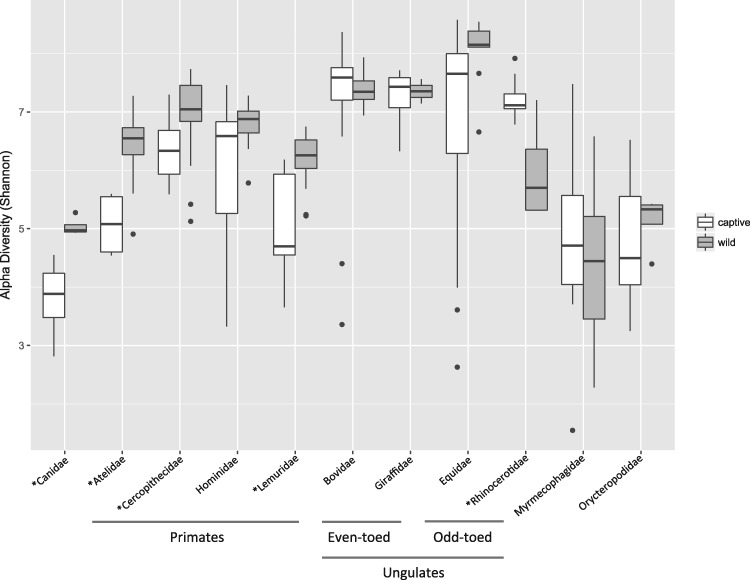
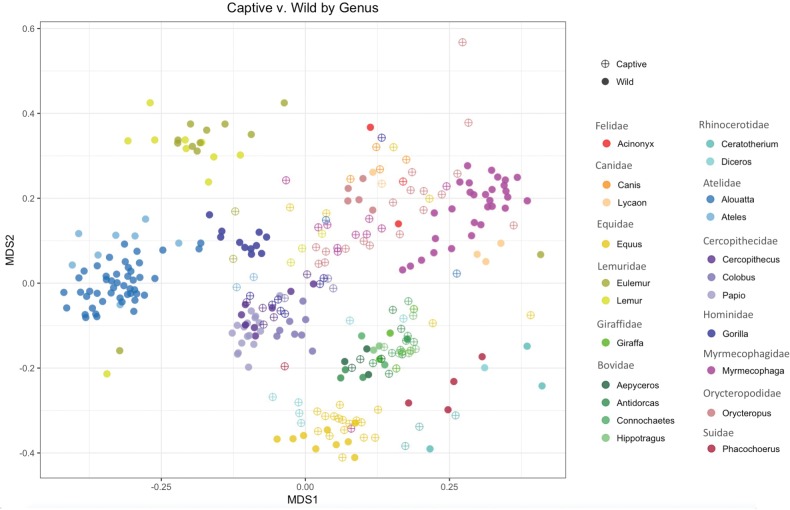
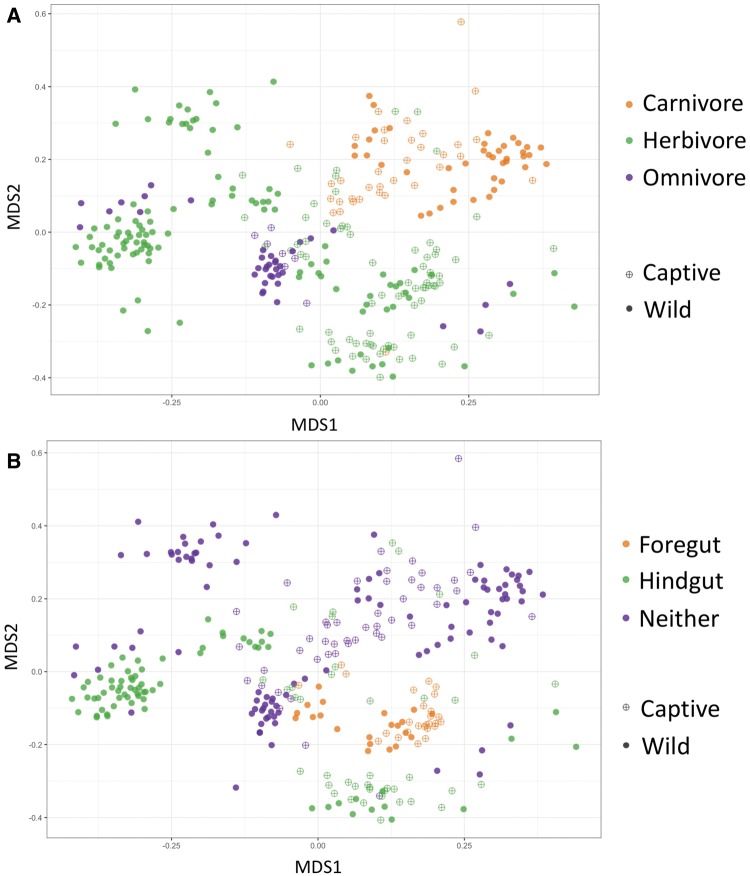
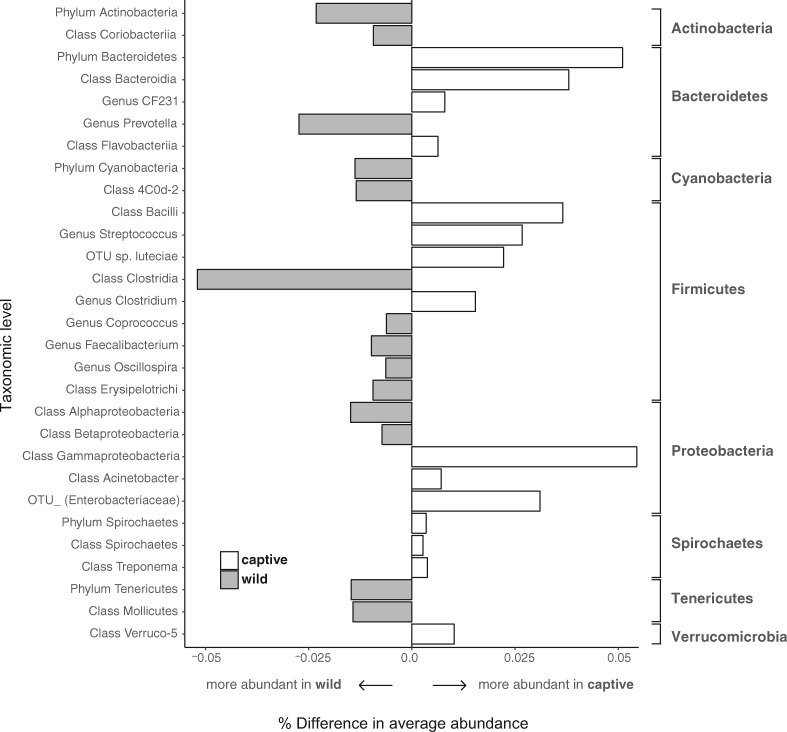
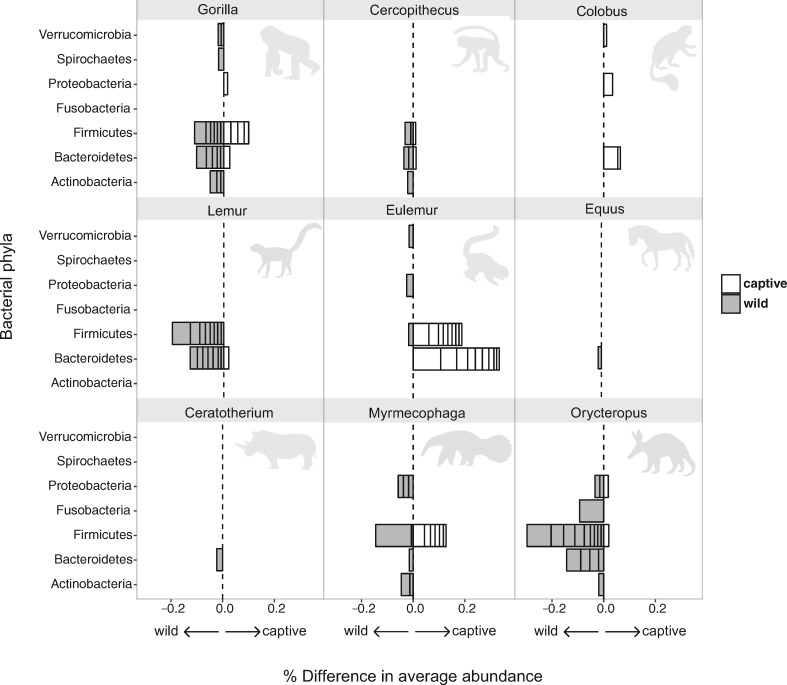
Recent studies increasingly note the effect of captivity or the built environment on the microbiome of humans and other animals. As symbiotic microbes are essential to many aspects of biology (e.g., digestive and immune functions), it is important to understand how lifestyle differences can impact the microbiome, and, consequently, the health of hosts. Animals living in captivity experience a range of changes that may influence the gut bacteria, such as diet changes, treatments, and reduced contact with other individuals, species and variable environmental substrates that act as sources of bacterial diversity. Thus far, initial results from previous studies point to a pattern of decreased bacterial diversity in captive animals. However, these studies are relatively limited in the scope of species that have been examined. Here we present a dataset that includes paired wild and captive samples from mammalian taxa across six Orders to investigate generalizable patterns of the effects captivity on mammalian gut bacteria. In comparing the wild to the captive condition, our results indicate that alpha diversity of the gut bacteria remains consistent in some mammalian hosts (bovids, giraffes, anteaters, and aardvarks), declines in the captive condition in some hosts (canids, primates, and equids), and increases in the captive condition in one host taxon (rhinoceros). Differences in gut bacterial beta diversity between the captive and wild state were observed for most of the taxa surveyed, except the even-toed ungulates (bovids and giraffes). Additionally, beta diversity variation was also strongly influenced by host taxonomic group, diet type, and gut fermentation physiology. Bacterial taxa that demonstrated larger shifts in relative abundance between the captive and wild states included members of the Firmicutes and Bacteroidetes. Overall, the patterns that we observe will inform a range of disciplines from veterinary practice to captive breeding efforts for biological conservation. Furthermore, bacterial taxa that persist in the captive state provide unique insight into symbiotic relationships with the host.
© The Author 2017. Published by Oxford University Press on behalf of the Society for Integrative and Comparative Biology. All rights reserved. For permissions please email: journals.permissions@oup.com.
Figures
References
-
- Amato KR, Van Belle S, Di Fiore A, Estrada A, Stumpf R, White B, Nelson KE, Knight R, Leigh SR.. 2017. Patterns in gut microbiota similarity associated with degree of sociality among sex classes of a neotropical primate. Microb Ecol 1–9. - PubMed
-
- Biagi E, Franceschi C, Rampelli S, Severgnini M, Ostan R, Turroni S, Consolandi C, Quercia S, Scurti M, Monti D, et al. 2016. Gut microbiota and extreme longevity. Curr Biol 26:1480–5. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
