

High-Throughput Approaches to Pinpoint Function within the Noncoding Genome
- PMID: 28985510
- PMCID: PMC5701515
- DOI: 10.1016/j.molcel.2017.09.017
High-Throughput Approaches to Pinpoint Function within the Noncoding Genome
Abstract
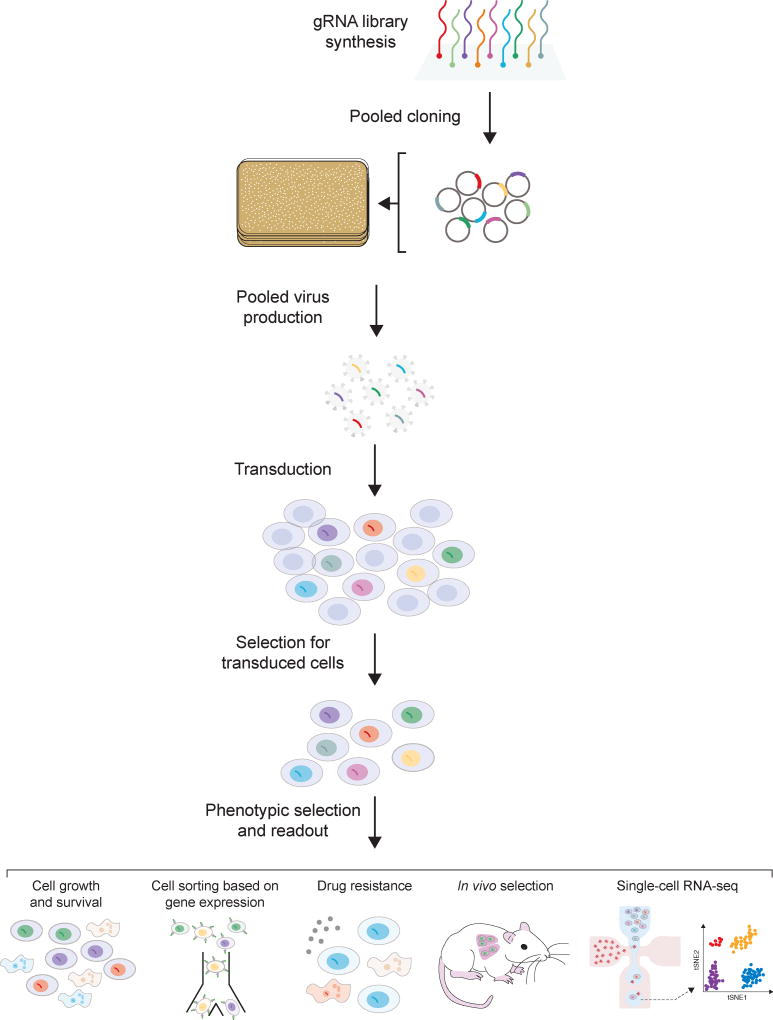
The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas nuclease system is a powerful tool for genome editing, and its simple programmability has enabled high-throughput genetic and epigenetic studies. These high-throughput approaches offer investigators a toolkit for functional interrogation of not only protein-coding genes but also noncoding DNA. Historically, noncoding DNA has lacked the detailed characterization that has been applied to protein-coding genes in large part because there has not been a robust set of methodologies for perturbing these regions. Although the majority of high-throughput CRISPR screens have focused on the coding genome to date, an increasing number of CRISPR screens targeting noncoding genomic regions continue to emerge. Here, we review high-throughput CRISPR-based approaches to uncover and understand functional elements within the noncoding genome and discuss practical aspects of noncoding library design and screen analysis.
Keywords: CRISPR; Cas9; conservation; enhancers; functional genomics; gene editing; gene expression; mutagenesis; noncoding genome; pooled screens.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures




Similar articles
-
Functional interrogation of non-coding DNA through CRISPR genome editing.Methods. 2017 May 15;121-122:118-129. doi: 10.1016/j.ymeth.2017.03.008. Epub 2017 Mar 10. Methods. 2017. PMID: 28288828 Free PMC article. Review.
-
Methods and Applications of CRISPR-Mediated Base Editing in Eukaryotic Genomes.Mol Cell. 2017 Oct 5;68(1):26-43. doi: 10.1016/j.molcel.2017.09.029. Mol Cell. 2017. PMID: 28985508 Free PMC article. Review.
-
Genome-scale CRISPR pooled screens.Anal Biochem. 2017 Sep 1;532:95-99. doi: 10.1016/j.ab.2016.05.014. Epub 2016 Jun 1. Anal Biochem. 2017. PMID: 27261176 Free PMC article.
-
CRISPR/Cas9 in Genome Editing and Beyond.Annu Rev Biochem. 2016 Jun 2;85:227-64. doi: 10.1146/annurev-biochem-060815-014607. Epub 2016 Apr 25. Annu Rev Biochem. 2016. PMID: 27145843 Review.
-
Conditional Control of CRISPR/Cas9 Function.Angew Chem Int Ed Engl. 2016 Apr 25;55(18):5394-9. doi: 10.1002/anie.201511441. Epub 2016 Mar 21. Angew Chem Int Ed Engl. 2016. PMID: 26996256 Review.
Cited by
-
Integrated design, execution, and analysis of arrayed and pooled CRISPR genome-editing experiments.Nat Protoc. 2018 May;13(5):946-986. doi: 10.1038/nprot.2018.005. Epub 2018 Apr 12. Nat Protoc. 2018. PMID: 29651054 Free PMC article.
-
Genomic annotation of disease-associated variants reveals shared functional contexts.Diabetologia. 2019 May;62(5):735-743. doi: 10.1007/s00125-019-4823-3. Epub 2019 Feb 12. Diabetologia. 2019. PMID: 30756131 Free PMC article. Review.
-
The non-coding genome in genetic brain disorders: new targets for therapy?Essays Biochem. 2021 Oct 27;65(4):671-683. doi: 10.1042/EBC20200121. Essays Biochem. 2021. PMID: 34414418 Free PMC article. Review.
-
Cancer CRISPR Screens In Vivo.Trends Cancer. 2018 May;4(5):349-358. doi: 10.1016/j.trecan.2018.03.002. Epub 2018 Mar 30. Trends Cancer. 2018. PMID: 29709259 Free PMC article. Review.
-
Integration of SNP Disease Association, eQTL, and Enrichment Analyses to Identify Risk SNPs and Susceptibility Genes in Chronic Obstructive Pulmonary Disease.Biomed Res Int. 2020 Dec 29;2020:3854196. doi: 10.1155/2020/3854196. eCollection 2020. Biomed Res Int. 2020. PMID: 33457407 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
