

Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage
- PMID: 28985763
- PMCID: PMC6389135
- DOI: 10.1186/s13059-017-1318-8
Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage
Abstract
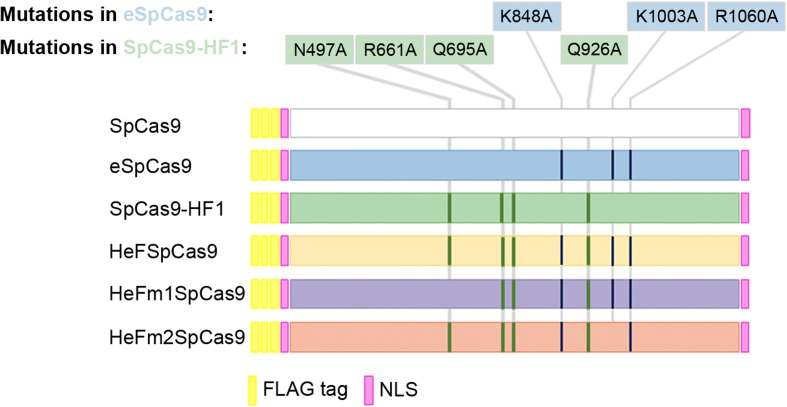
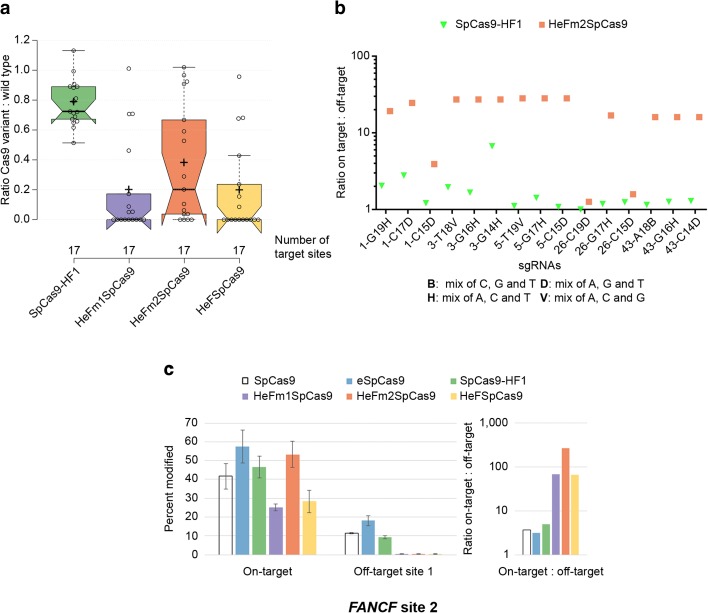
Background: The propensity for off-target activity of Streptococcus pyogenes Cas9 (SpCas9) has been considerably decreased by rationally engineered variants with increased fidelity (eSpCas9; SpCas9-HF1). However, a subset of targets still generate considerable off-target effects. To deal specifically with these targets, we generated new "Highly enhanced Fidelity" nuclease variants (HeFSpCas9s) containing mutations from both eSpCas9 and SpCas9-HF1 and examined these improved nuclease variants side by side to decipher the factors that affect their specificities and to determine the optimal nuclease for applications sensitive to off-target effects.
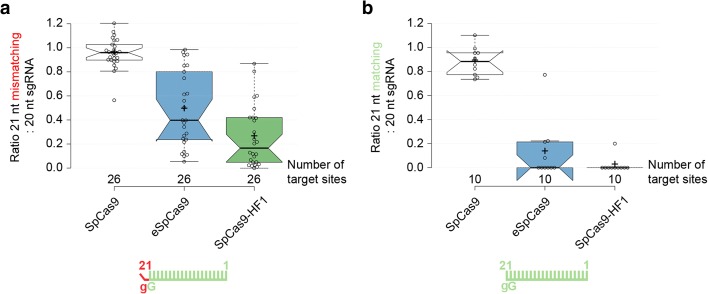
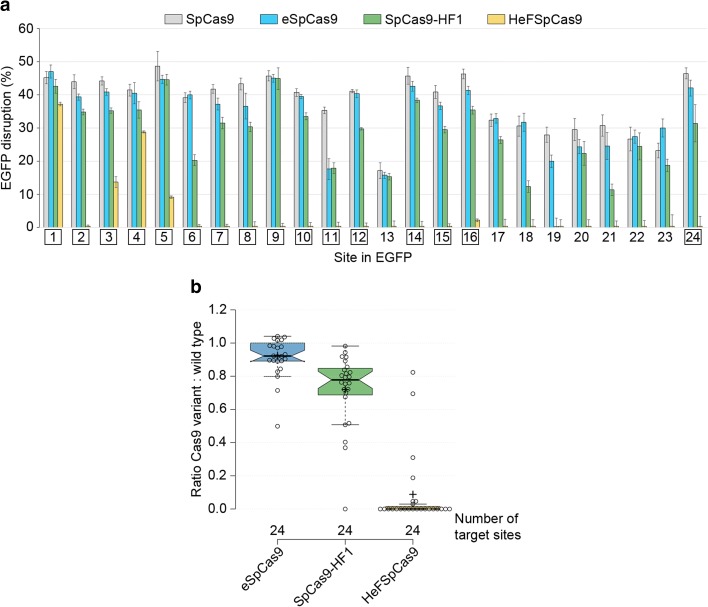
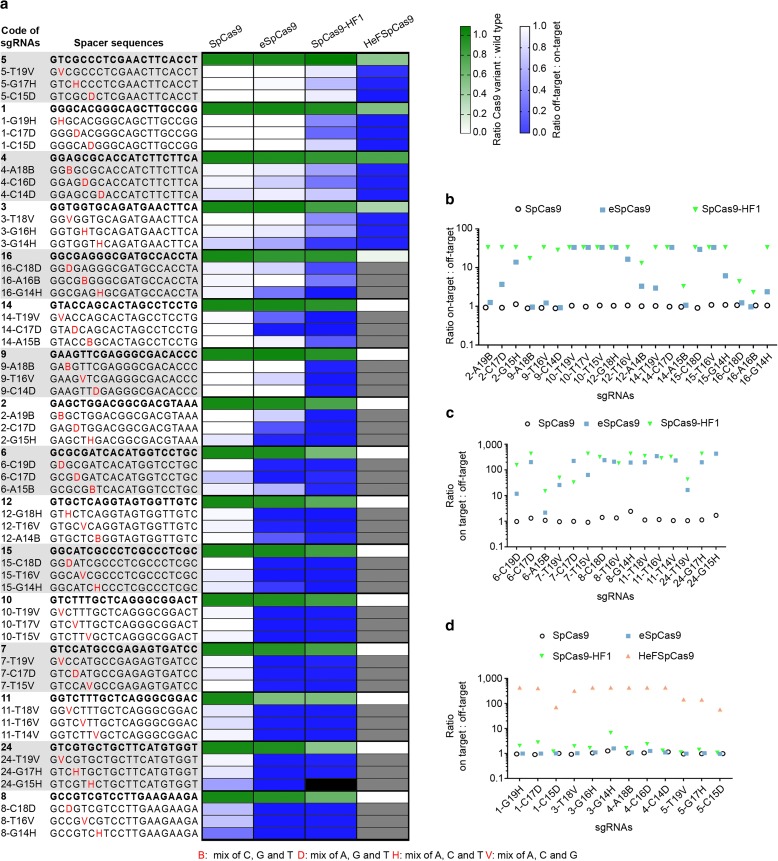
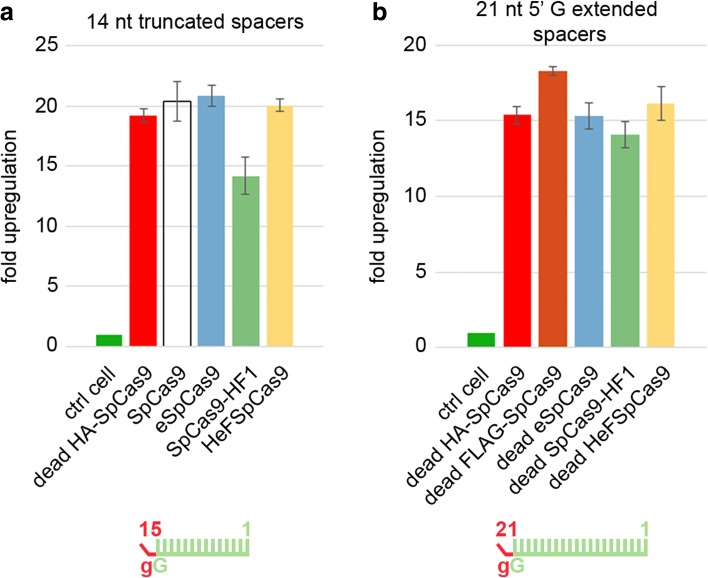
Results: These three increased-fidelity nucleases can routinely be used only with perfectly matching 20-nucleotide-long spacers, a matching 5' G extension being more detrimental to their activities than a mismatching one. HeFSpCas9 exhibit substantially improved specificity for those targets for which eSpCas9 and SpCas9-HF1 have higher off-target propensity. The targets can also be ranked by their cleavability and off-target effects manifested by the increased fidelity nucleases. Furthermore, we show that the mutations in these variants may diminish the cleavage, but not the DNA-binding, of SpCas9s.
Conclusions: No single nuclease variant shows generally superior fidelity; instead, for highest specificity cleavage, each target needs to be matched with an appropriate high-fidelity nuclease. We provide here a framework for generating new nuclease variants for targets that currently have no matching optimal nuclease, and offer a simple means for identifying the optimal nuclease for targets in the absence of accurate target-ranking prediction tools.
Keywords: CRISPR; Cas9; Disruption assay; Extended sgRNA; HeFSpCas9; HeFm2SpCas9; High fidelity nuclease; Off-target; SpCas9-HF1; Truncated sgRNA; eSpCas9; sgRNA.
Conflict of interest statement
Ethics approval and consent to participate
Ethics approval was not needed for the study.
Competing interests
The authors declare that they have no significant competing financial, professional, or personal interests that might have influenced the performance or presentation of the work described in this manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
