

Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer's Disease
- PMID: 28988799
- PMCID: PMC5748263
- DOI: 10.1016/j.freeradbiomed.2017.10.001
Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer's Disease
Abstract
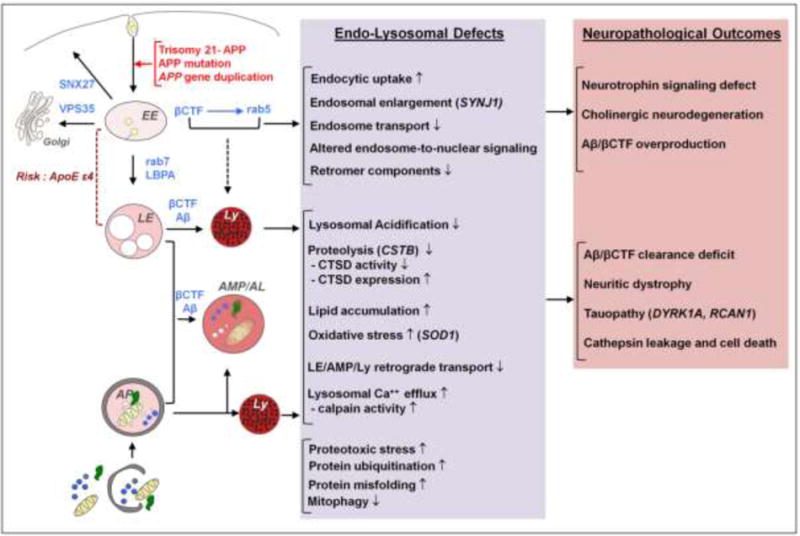
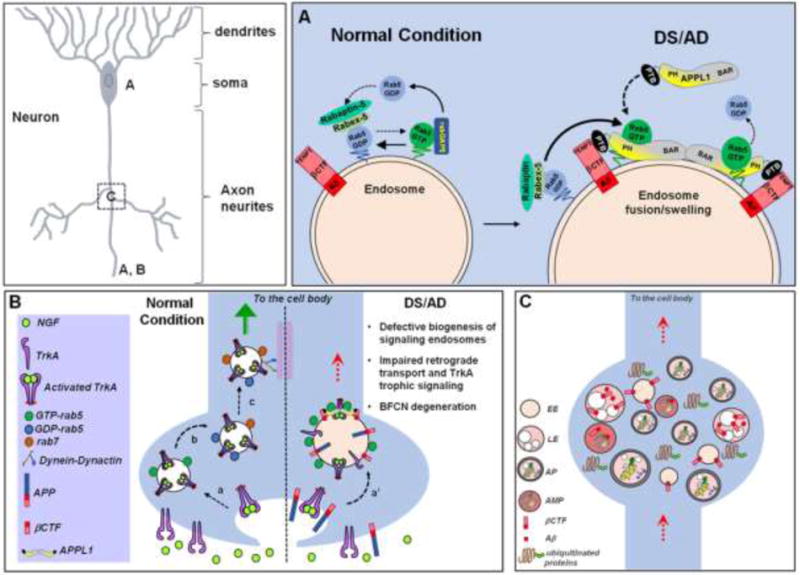
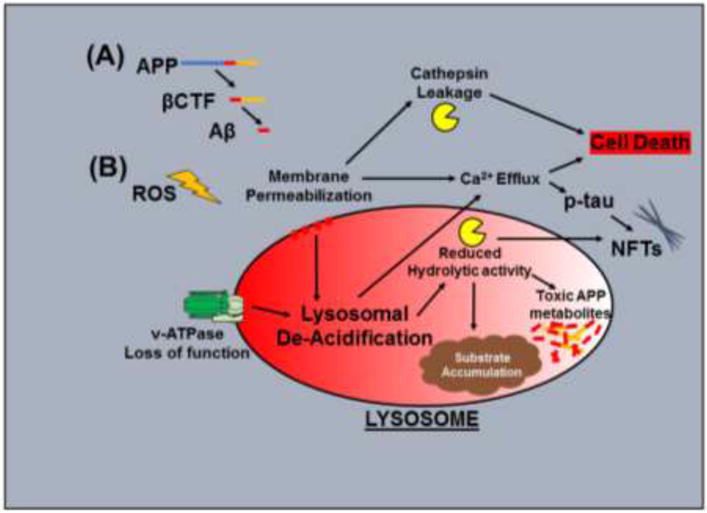
Individuals with Down syndrome (DS) have an increased risk of early-onset Alzheimer's Disease (AD), largely owing to a triplication of the APP gene, located on chromosome 21. In DS and AD, defects in endocytosis and lysosomal function appear at the earliest stages of disease development and progress to widespread failure of intraneuronal waste clearance, neuritic dystrophy and neuronal cell death. The same genetic factors that cause or increase AD risk are also direct causes of endosomal-lysosomal dysfunction, underscoring the essential partnership between this dysfunction and APP metabolites in AD pathogenesis. The appearance of APP-dependent endosome anomalies in DS beginning in infancy and evolving into the full range of AD-related endosomal-lysosomal deficits provides a unique opportunity to characterize the earliest pathobiology of AD preceding the classical neuropathological hallmarks. Facilitating this characterization is the authentic recapitulation of this endosomal pathobiology in peripheral cells from people with DS and in trisomy mouse models. Here, we review current research on endocytic-lysosomal dysfunction in DS and AD, the emerging importance of APP/βCTF in initiating this dysfunction, and the potential roles of additional trisomy 21 genes in accelerating endosomal-lysosomal impairment in DS. Collectively, these studies underscore the growing value of investigating DS to probe the biological origins of AD as well as to understand and ameliorate the developmental disability of DS.
Keywords: Alzheimer's Disease; Autophagy; Down syndrome; Endosomes; Lysosomes.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Arai Y, Ijuin T, Takenawa T, Becker LE, Takashima S. Excessive expression of synaptojanin in brains with Down syndrome. Brain Dev. 2002;24:67–72. - PubMed
-
- Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013;288:1295–1306. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
