

Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands
- PMID: 28991257
- PMCID: PMC5675000
- DOI: 10.1038/ng.3970
Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands
Abstract
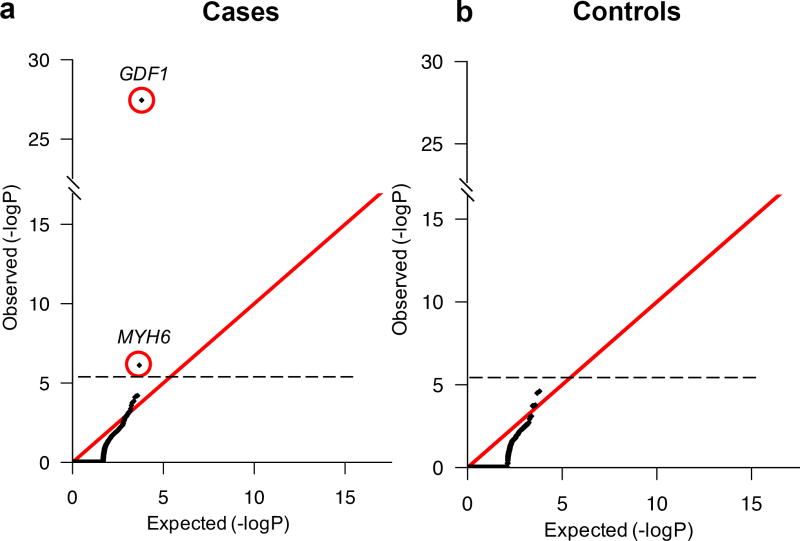
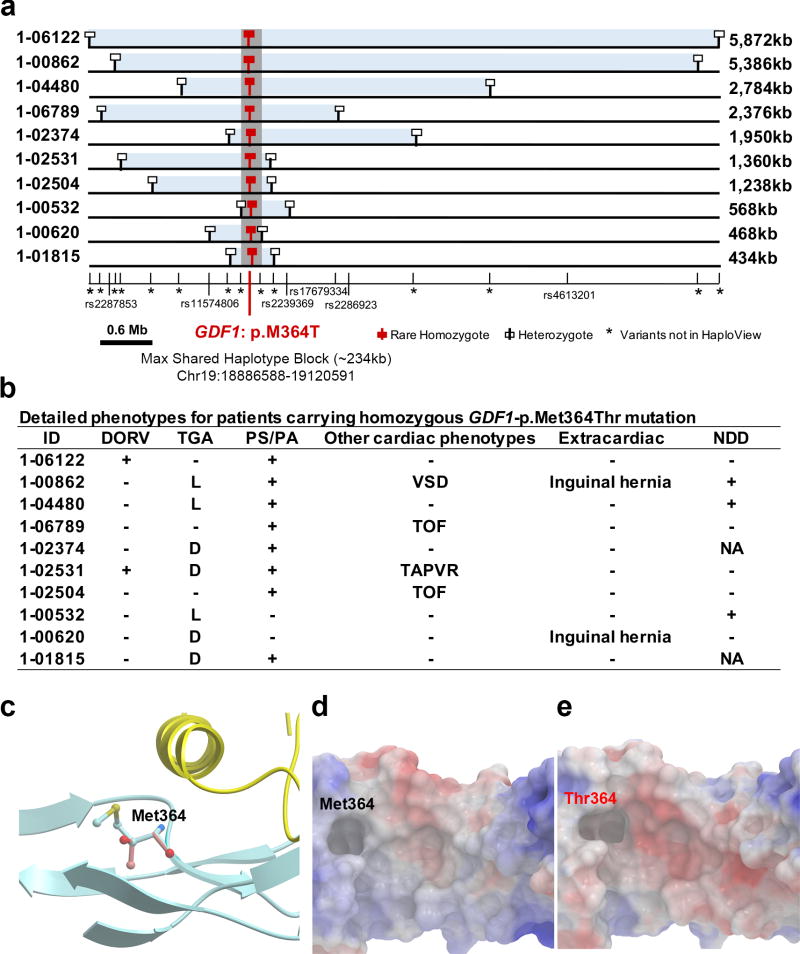
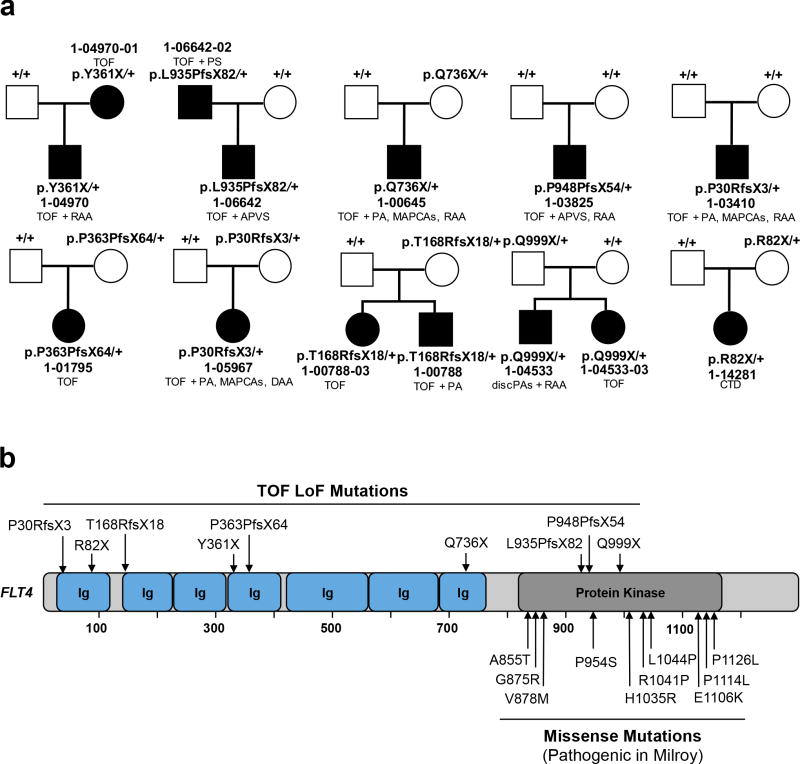
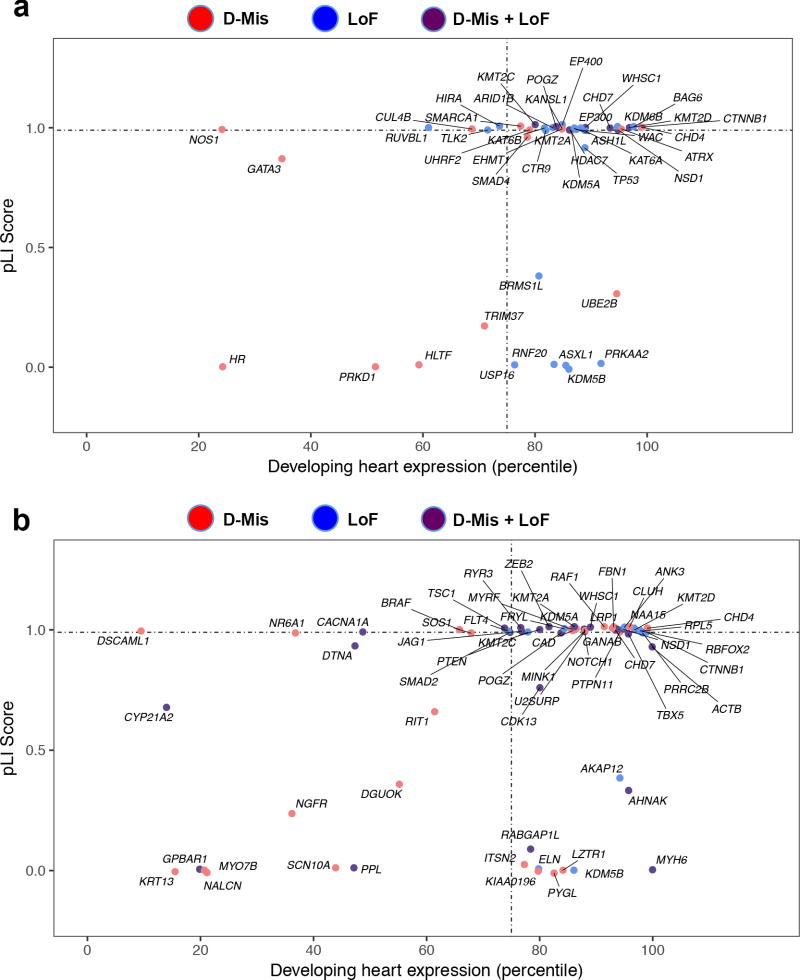
Congenital heart disease (CHD) is the leading cause of mortality from birth defects. Here, exome sequencing of a single cohort of 2,871 CHD probands, including 2,645 parent-offspring trios, implicated rare inherited mutations in 1.8%, including a recessive founder mutation in GDF1 accounting for ∼5% of severe CHD in Ashkenazim, recessive genotypes in MYH6 accounting for ∼11% of Shone complex, and dominant FLT4 mutations accounting for 2.3% of Tetralogy of Fallot. De novo mutations (DNMs) accounted for 8% of cases, including ∼3% of isolated CHD patients and ∼28% with both neurodevelopmental and extra-cardiac congenital anomalies. Seven genes surpassed thresholds for genome-wide significance, and 12 genes not previously implicated in CHD had >70% probability of being disease related. DNMs in ∼440 genes were inferred to contribute to CHD. Striking overlap between genes with damaging DNMs in probands with CHD and autism was also found.
Conflict of interest statement
None
Figures
Comment in
-
The new era of whole-exome sequencing in congenital heart disease: brand-new insights into rare pathogenic variants.J Thorac Dis. 2018 Jun;10(Suppl 17):S1923-S1929. doi: 10.21037/jtd.2018.05.56. J Thorac Dis. 2018. PMID: 30023082 Free PMC article. No abstract available.
References
-
- van der Linde D, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241–7. - PubMed
-
- Marino BS, et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012;126:1143–72. - PubMed
MeSH terms
Substances
Grants and funding
- UM1 HL098123/HL/NHLBI NIH HHS/United States
- U01 HL098162/HL/NHLBI NIH HHS/United States
- S10 OD018522/OD/NIH HHS/United States
- R01 GM104390/GM/NIGMS NIH HHS/United States
- R01 HL132024/HL/NHLBI NIH HHS/United States
- UM1 HL128711/HL/NHLBI NIH HHS/United States
- UM1 HL098147/HL/NHLBI NIH HHS/United States
- U01 HL098147/HL/NHLBI NIH HHS/United States
- UL1 TR001067/TR/NCATS NIH HHS/United States
- U01 HL098163/HL/NHLBI NIH HHS/United States
- UM1 HL098162/HL/NHLBI NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- U01 HL098188/HL/NHLBI NIH HHS/United States
- T32 HD007149/HD/NICHD NIH HHS/United States
- S10 OD018521/OD/NIH HHS/United States
- U01 HL098153/HL/NHLBI NIH HHS/United States
- U01 HL131003/HL/NHLBI NIH HHS/United States
- U54 HG006504/HG/NHGRI NIH HHS/United States
- U01 HL098180/HL/NHLBI NIH HHS/United States
- UL1 TR000003/TR/NCATS NIH HHS/United States
- UM1 HL098160/HL/NHLBI NIH HHS/United States
- T32 GM007205/GM/NIGMS NIH HHS/United States
- U01 HL098160/HL/NHLBI NIH HHS/United States
- U01 HL098123/HL/NHLBI NIH HHS/United States
- U54 HD090255/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
