

Applications of sequence coevolution in membrane protein biochemistry
- PMID: 28993150
- PMCID: PMC5807202
- DOI: 10.1016/j.bbamem.2017.10.004
Applications of sequence coevolution in membrane protein biochemistry
Abstract
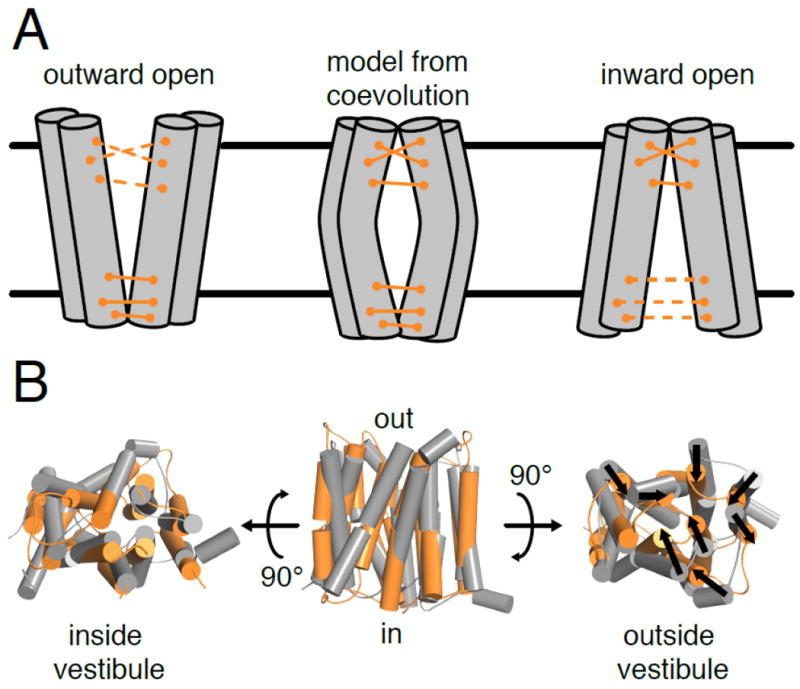
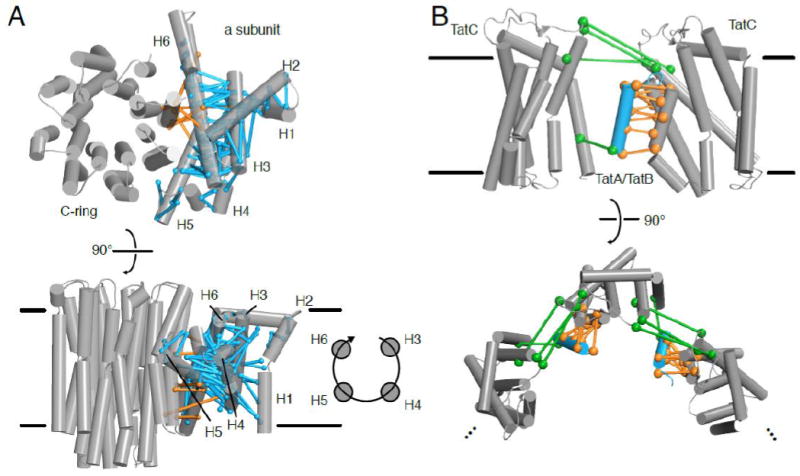
Recently, protein sequence coevolution analysis has matured into a predictive powerhouse for protein structure and function. Direct methods, which use global statistical models of sequence coevolution, have enabled the prediction of membrane and disordered protein structures, protein complex architectures, and the functional effects of mutations in proteins. The field of membrane protein biochemistry and structural biology has embraced these computational techniques, which provide functional and structural information in an otherwise experimentally-challenging field. Here we review recent applications of protein sequence coevolution analysis to membrane protein structure and function and highlight the promising directions and future obstacles in these fields. We provide insights and guidelines for membrane protein biochemists who wish to apply sequence coevolution analysis to a given experimental system.
Keywords: ABC transporters; Clustered protocadherin; Conformational changes; Membrane proteins; Protein-protein interactions; Sequence coevolution analysis.
Copyright © 2017 Elsevier B.V. All rights reserved.
Figures
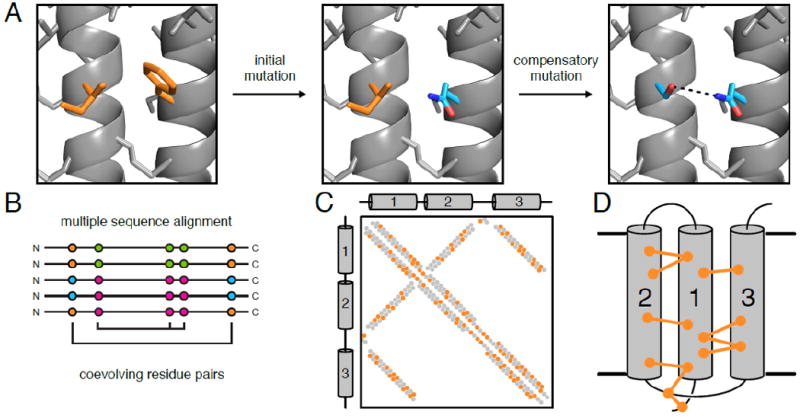
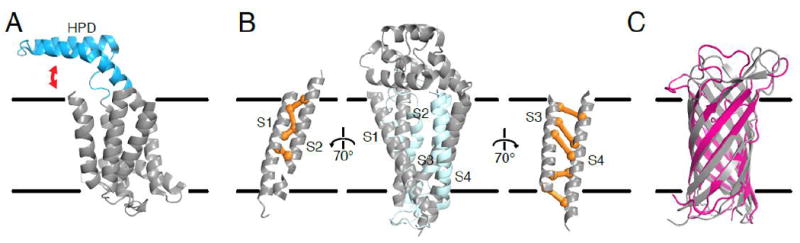
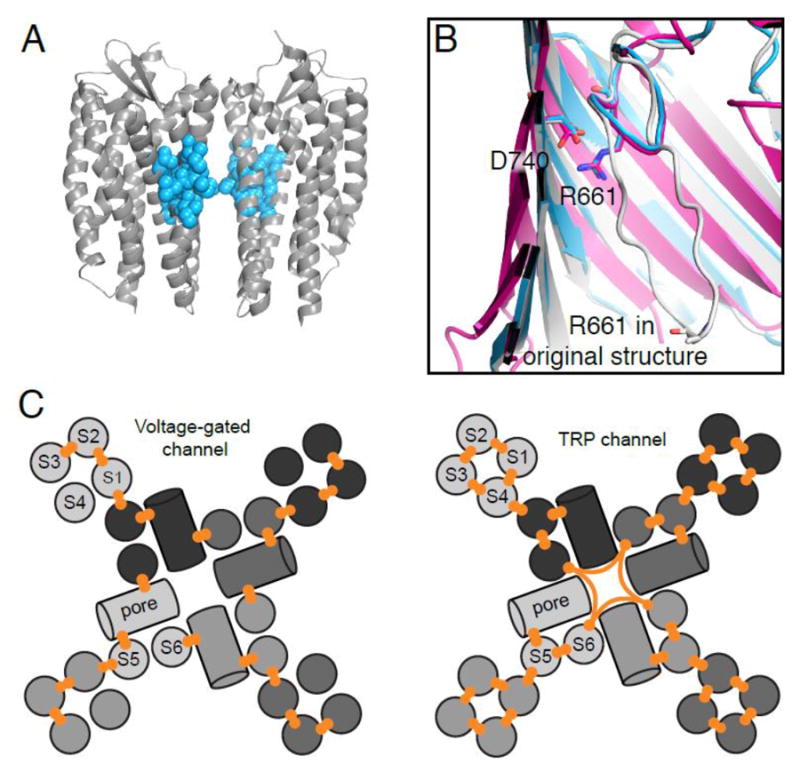
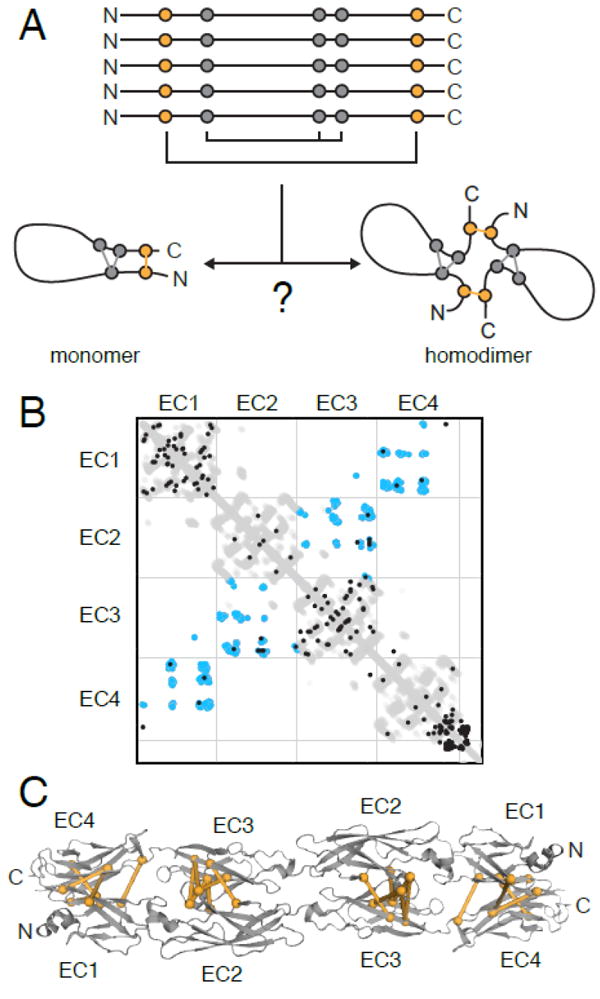
References
-
- Göbel U, Sander C, Schneider R, Valencia A. Correlated Mutations and Residue Contacts in Proteins. Proteins Struct Funct Genet. 1994;18:309–317. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
