

Effect of Mutant p53 Proteins on Glycolysis and Mitochondrial Metabolism
- PMID: 28993478
- PMCID: PMC5705820
- DOI: 10.1128/MCB.00328-17
Effect of Mutant p53 Proteins on Glycolysis and Mitochondrial Metabolism
Abstract
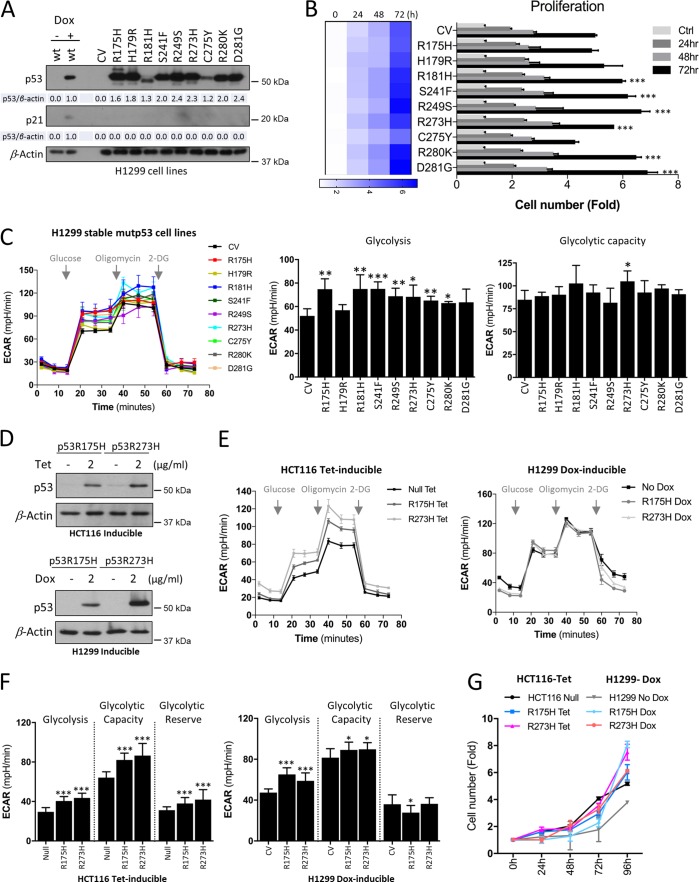
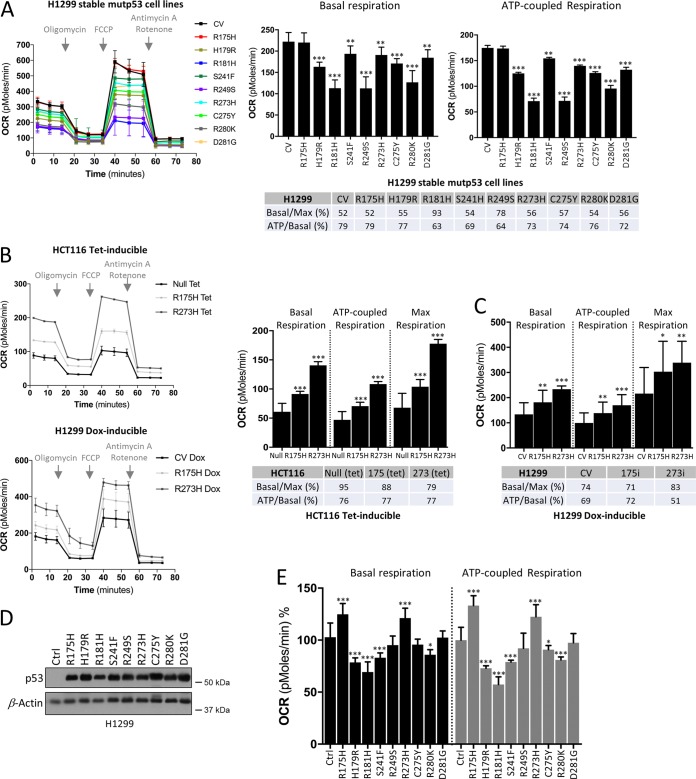
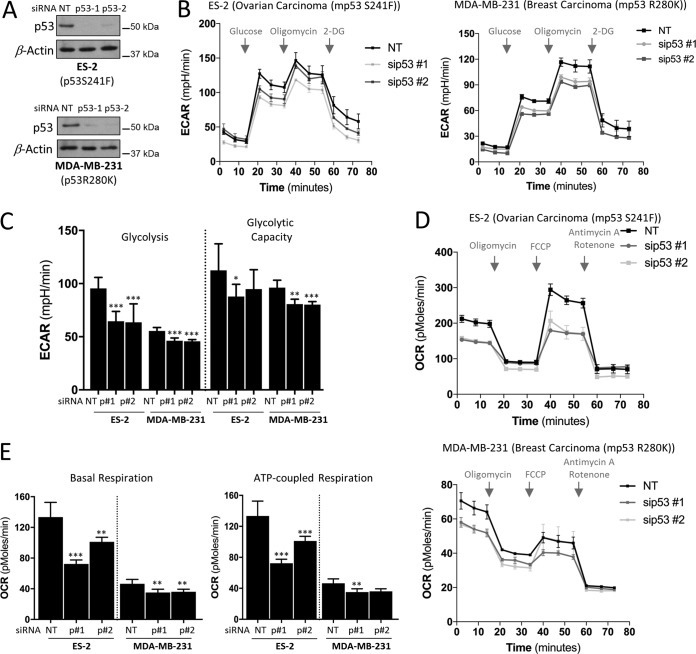
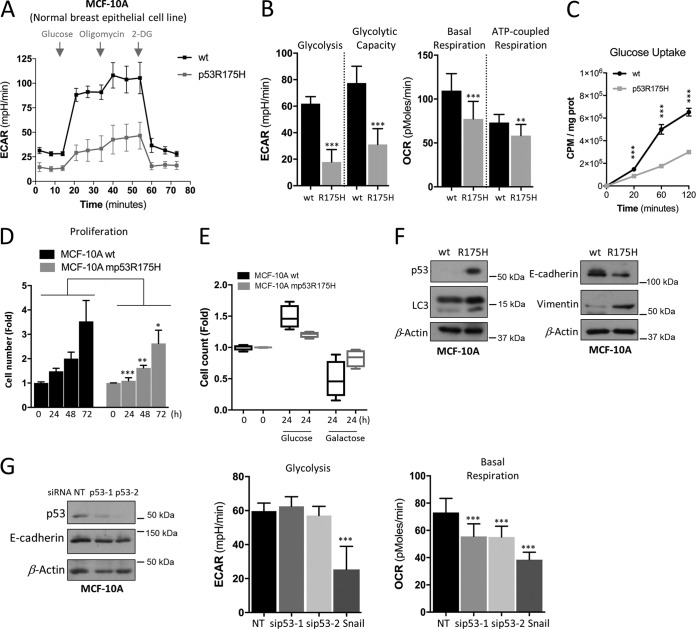
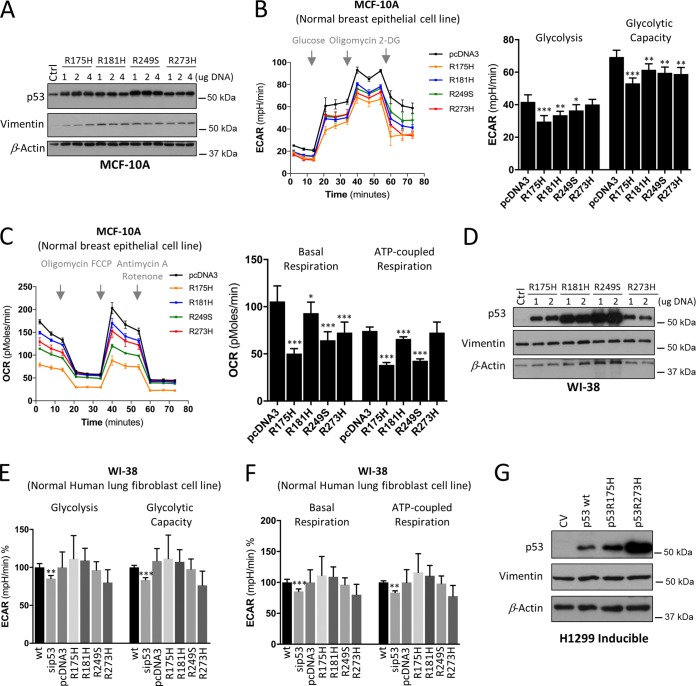
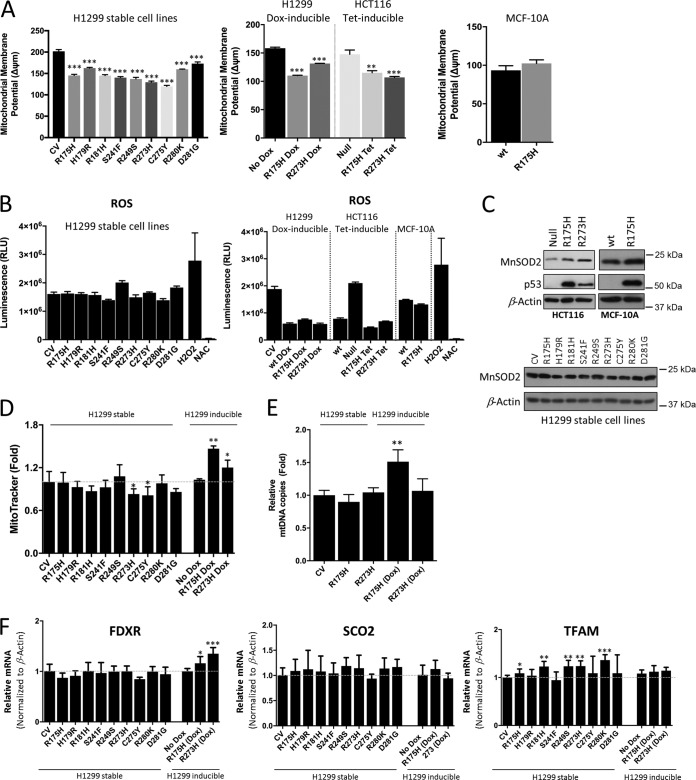
TP53 is one of the most commonly mutated genes in human cancers. Unlike other tumor suppressors that are frequently deleted or acquire loss-of-function mutations, the majority of TP53 mutations in tumors are missense substitutions, which lead to the expression of full-length mutant proteins that accumulate in cancer cells and may confer unique gain-of-function (GOF) activities to promote tumorigenic events. Recently, mutant p53 proteins have been shown to mediate metabolic changes as a novel GOF to promote tumor development. There is a strong rationale that the GOF activities, including alterations in cellular metabolism, might vary between the different p53 mutants. Accordingly, the effect of different mutant p53 proteins on cancer cell metabolism is largely unknown. In this study, we have metabolically profiled several individual frequently occurring p53 mutants in cancers, focusing on glycolytic and mitochondrial oxidative phosphorylation pathways. Our investigation highlights the diversity of different p53 mutants in terms of their effect on metabolism, which might provide a foundation for the development of more effective targeted pharmacological approaches toward variants of mutant p53.
Keywords: EMT; OxPhos; cancer; glycolysis; metabolism; mutant p53.
Copyright © 2017 American Society for Microbiology.
Figures
References
-
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. 1994. Tumor spectrum analysis in p53-mutant mice. Curr Biol 4:1–7. - PubMed
-
- Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, Cromer A, Brugge JS, Sansom OJ, Norman JC, Vousden KH. 2009. Mutant p53 drives invasion by promoting integrin recycling. Cell 139:1327–1341. doi: 10.1016/j.cell.2009.11.026. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous