

Factor H Competitor Generated by Gene Conversion Events Associates with Atypical Hemolytic Uremic Syndrome
- PMID: 28993505
- PMCID: PMC5748918
- DOI: 10.1681/ASN.2017050518
Factor H Competitor Generated by Gene Conversion Events Associates with Atypical Hemolytic Uremic Syndrome
Abstract
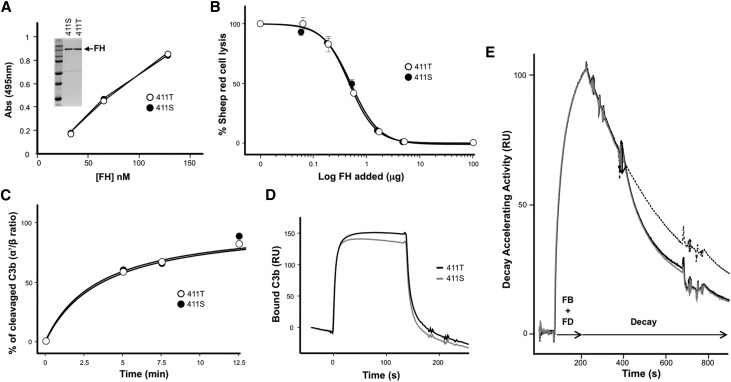
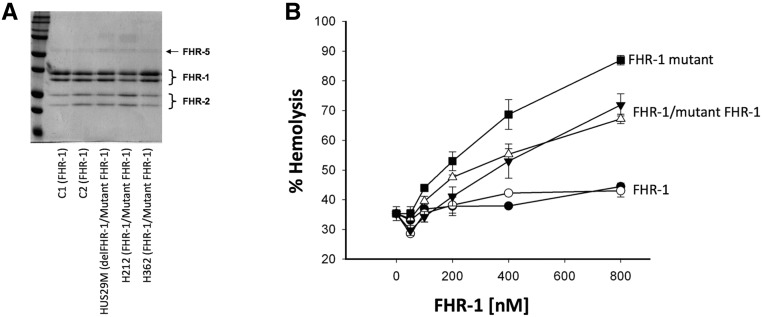
Atypical hemolytic uremic syndrome (aHUS), a rare form of thrombotic microangiopathy caused by complement pathogenic variants, mainly affects the kidney microvasculature. A retrospective genetic analysis in our aHUS cohort (n=513) using multiple ligation probe amplification uncovered nine unrelated patients carrying a genetic abnormality in the complement factor H related 1 gene (CFHR1) that originates by recurrent gene conversion events between the CFH and CFHR1 genes. The novel CFHR1 mutants encode an FHR-1 protein with two amino acid substitutions, L290S and A296V, converting the FHR-1 C terminus into that of factor H (FH). Next-generation massive-parallel DNA sequencing (NGS) analysis did not detect these genetic abnormalities. In addition to the CFHR1 mutant, six patients carried the previously uncharacterized CFH-411T variant. In functional analyses, the mutant FHR-1 protein strongly competed the binding of FH to cell surfaces, impairing complement regulation, whereas the CFH-411T polymorphism lacked functional consequences. Carriers of the CFHR1 mutation presented with severe aHUS during adulthood; 57% of affected women in this cohort presented during the postpartum period. Analyses in patients and unaffected carriers showed that FH plasma levels determined by the nonmutated chromosome modulate disease penetrance. Crucially, in the activated endothelial (HMEC-1) cell assay, reduced FH plasma levels produced by the nonmutated chromosome correlated inversely with impairment of complement regulation, measured as C5b-9 deposition. Our data advance understanding of the genetic complexities underlying aHUS, illustrate the importance of performing functional analysis, and support the use of complementary assays to disclose genetic abnormalities not revealed by current NGS analysis.
Keywords: CFHR1; complement; gene conversion; hemolytic uremic syndrome.
Copyright © 2018 by the American Society of Nephrology.
Figures





References
-
- Moake JL: Thrombotic microangiopathies. N Engl J Med 347: 589–600, 2002 - PubMed
-
- Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Fremeaux-Bacchi V, Kavanagh D, Nester CM, Noris M, Pickering MC, Rodriguez de Cordoba S, Roumenina LT, Sethi S, Smith RJ; Conference P : Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 91: 539–551, 2017 - PubMed
-
- de Córdoba SR: Complement genetics and susceptibility to inflammatory disease. Lessons from genotype-phenotype correlations. Immunobiology 221: 709–714, 2016 - PubMed
-
- Francis NJ, McNicholas B, Awan A, Waldron M, Reddan D, Sadlier D, Kavanagh D, Strain L, Marchbank KJ, Harris CL, Goodship THJ: A novel hybrid CFH/CFHR3 gene generated by a microhomology-mediated deletion in familial atypical hemolytic uremic syndrome. Blood 119: 591–601, 2012 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
