

Geometry Optimization with Machine Trained Topological Atoms
- PMID: 28993674
- PMCID: PMC5634454
- DOI: 10.1038/s41598-017-12600-3
Geometry Optimization with Machine Trained Topological Atoms
Abstract
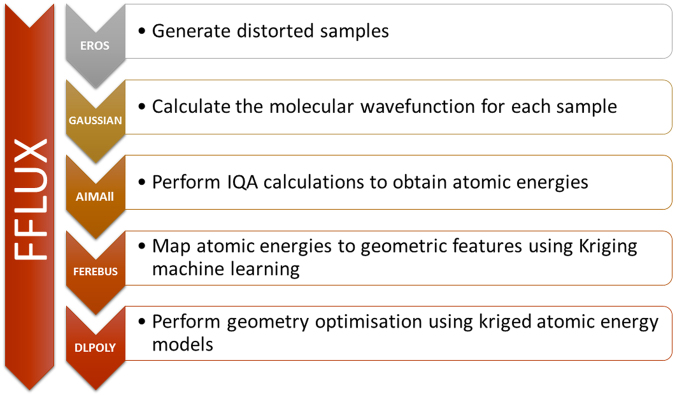
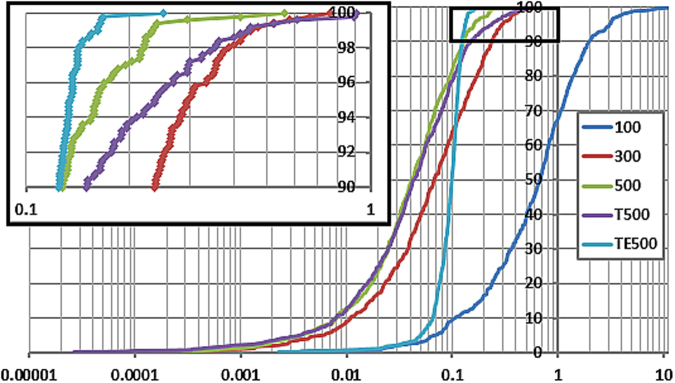
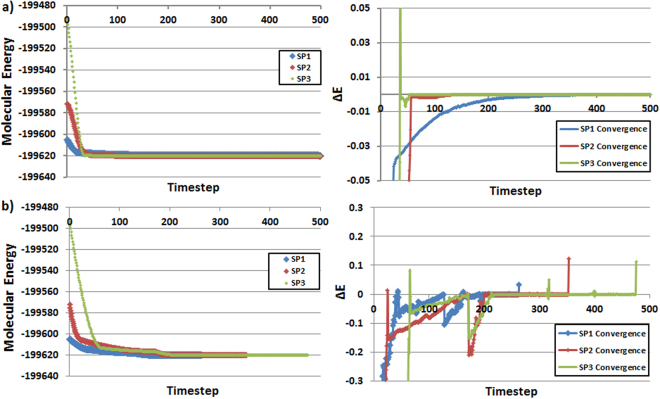
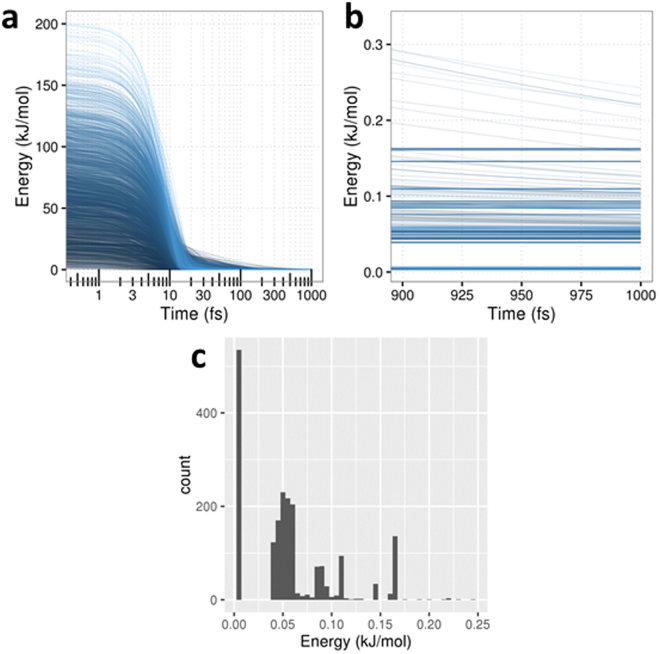
The geometry optimization of a water molecule with a novel type of energy function called FFLUX is presented, which bypasses the traditional bonded potentials. Instead, topologically-partitioned atomic energies are trained by the machine learning method kriging to predict their IQA atomic energies for a previously unseen molecular geometry. Proof-of-concept that FFLUX's architecture is suitable for geometry optimization is rigorously demonstrated. It is found that accurate kriging models can optimize 2000 distorted geometries to within 0.28 kJ mol-1 of the corresponding ab initio energy, and 50% of those to within 0.05 kJ mol-1. Kriging models are robust enough to optimize the molecular geometry to sub-noise accuracy, when two thirds of the geometric inputs are outside the training range of that model. Finally, the individual components of the potential energy are analyzed, and chemical intuition is reflected in the independent behavior of the three energy terms [Formula: see text](intra-atomic), [Formula: see text] (electrostatic) and [Formula: see text] (exchange), in contrast to standard force fields.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
The accuracy of ab initio calculations without ab initio calculations for charged systems: Kriging predictions of atomistic properties for ions in aqueous solutions.J Chem Phys. 2018 Jun 28;148(24):241724. doi: 10.1063/1.5022174. J Chem Phys. 2018. PMID: 29960379
-
Description of Potential Energy Surfaces of Molecules Using FFLUX Machine Learning Models.J Chem Theory Comput. 2019 Jan 8;15(1):116-126. doi: 10.1021/acs.jctc.8b00806. Epub 2018 Dec 3. J Chem Theory Comput. 2019. PMID: 30507180
-
Prediction of Intramolecular Polarization of Aromatic Amino Acids Using Kriging Machine Learning.J Chem Theory Comput. 2014 Sep 9;10(9):3708-19. doi: 10.1021/ct500416k. J Chem Theory Comput. 2014. PMID: 26588516
-
Multipolar Electrostatic Energy Prediction for all 20 Natural Amino Acids Using Kriging Machine Learning.J Chem Theory Comput. 2016 Jun 14;12(6):2742-51. doi: 10.1021/acs.jctc.6b00457. Epub 2016 Jun 3. J Chem Theory Comput. 2016. PMID: 27224739
-
Calculations on noncovalent interactions and databases of benchmark interaction energies.Acc Chem Res. 2012 Apr 17;45(4):663-72. doi: 10.1021/ar200255p. Epub 2012 Jan 6. Acc Chem Res. 2012. PMID: 22225511 Review.
Cited by
-
Explainable chemical artificial intelligence from accurate machine learning of real-space chemical descriptors.Nat Commun. 2024 May 21;15(1):4345. doi: 10.1038/s41467-024-48567-9. Nat Commun. 2024. PMID: 38773090 Free PMC article.
-
A FFLUX Water Model: Flexible, Polarizable and with a Multipolar Description of Electrostatics.J Comput Chem. 2020 Mar 15;41(7):619-628. doi: 10.1002/jcc.26111. Epub 2019 Nov 20. J Comput Chem. 2020. PMID: 31747059 Free PMC article.
-
Interacting Quantum Atoms-A Review.Molecules. 2020 Sep 3;25(17):4028. doi: 10.3390/molecules25174028. Molecules. 2020. PMID: 32899346 Free PMC article. Review.
-
Choosing the right molecular machine learning potential.Chem Sci. 2021 Sep 15;12(43):14396-14413. doi: 10.1039/d1sc03564a. eCollection 2021 Nov 10. Chem Sci. 2021. PMID: 34880991 Free PMC article.
-
Accelerating explicit solvent models of heterogeneous catalysts with machine learning interatomic potentials.Chem Sci. 2023 Jul 12;14(31):8338-8354. doi: 10.1039/d3sc02482b. eCollection 2023 Aug 9. Chem Sci. 2023. PMID: 37564405 Free PMC article.
References
-
- Bader, R. F. W. Atoms in Molecules. A Quantum Theory. (Oxford Univ. Press, Oxford, Great Britain, 1990).
-
- Popelier, P. L. A. The Quantum Theory of Atoms in Molecules. In The Nature of the Chemical Bond Revisited (eds Frenking, G. & Shaik, S.) 271–308 (Wiley-VCH, Chapter 8, 2014).
-
- Matta, C. F. & Boyd, R. J. The Quantum Theory of Atoms in Molecules. From Solid State to DNA and Drug Design. (Wiley-VCH, Weinheim, Germany, 2007).
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
