

Matrix metalloproteinase-2 knockout prevents angiotensin II-induced vascular injury
- PMID: 29016715
- PMCID: PMC5852628
- DOI: 10.1093/cvr/cvx115
Matrix metalloproteinase-2 knockout prevents angiotensin II-induced vascular injury
Abstract
Aims: Matrix metalloproteinases (MMPs) have been implicated in the development of hypertension in animal models and humans. Mmp2 deletion did not change Ang II-induced blood pressure (BP) rise. However, whether Mmp2 knockout affects angiotensin (Ang) II-induced vascular injury has not been tested. We sought to determine whether Mmp2 knockout will prevent Ang II-induced vascular injury.
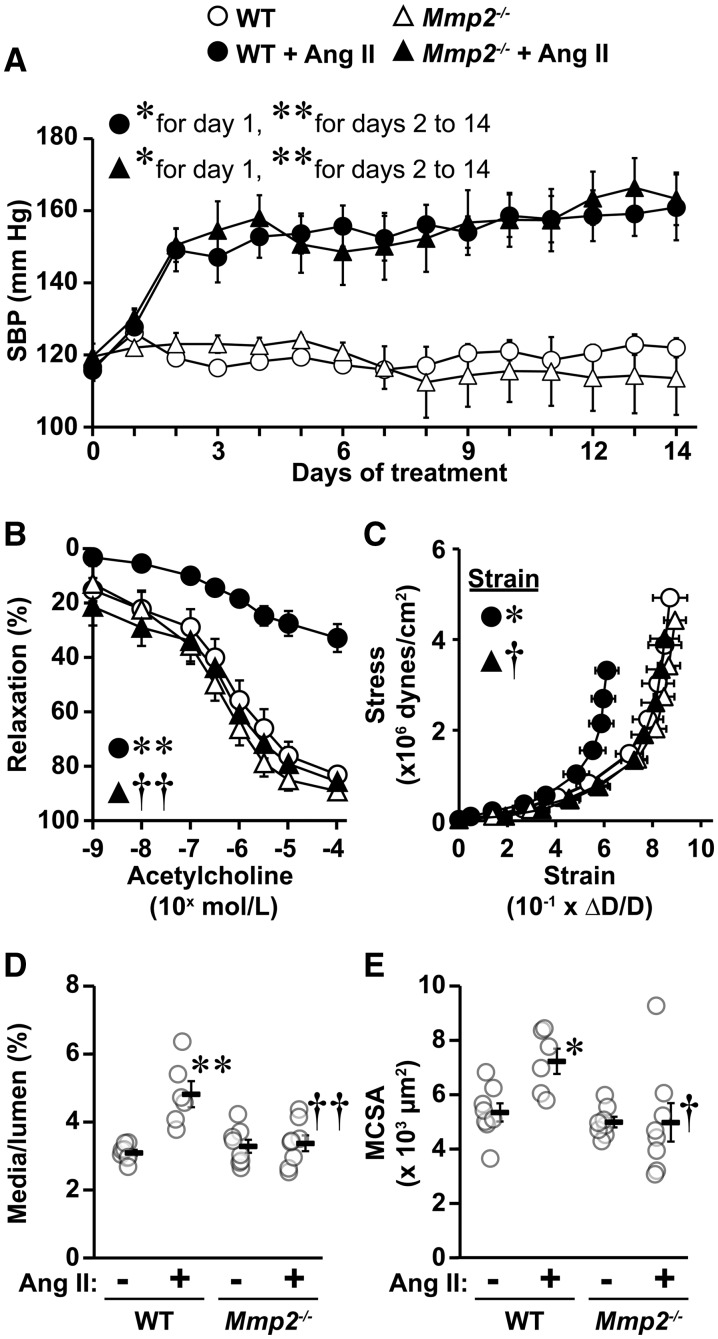
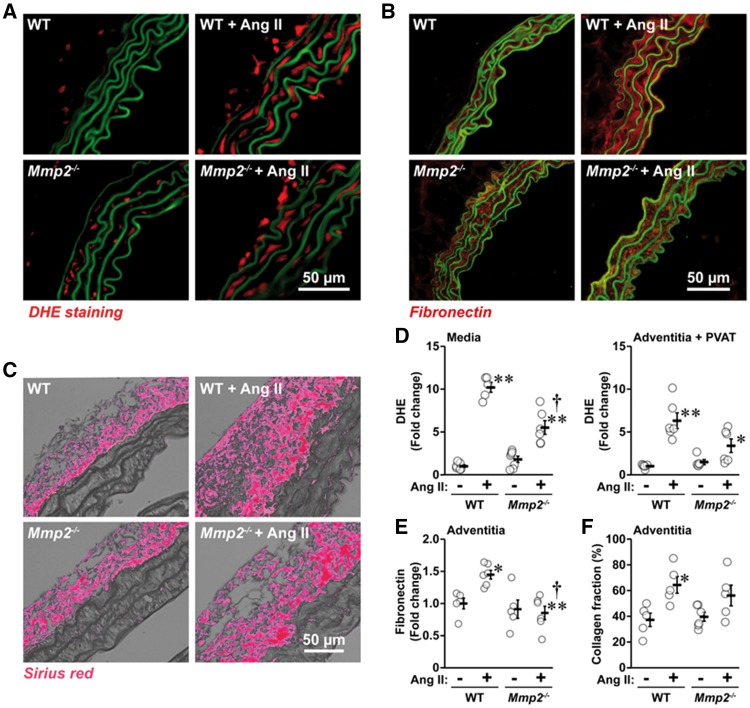
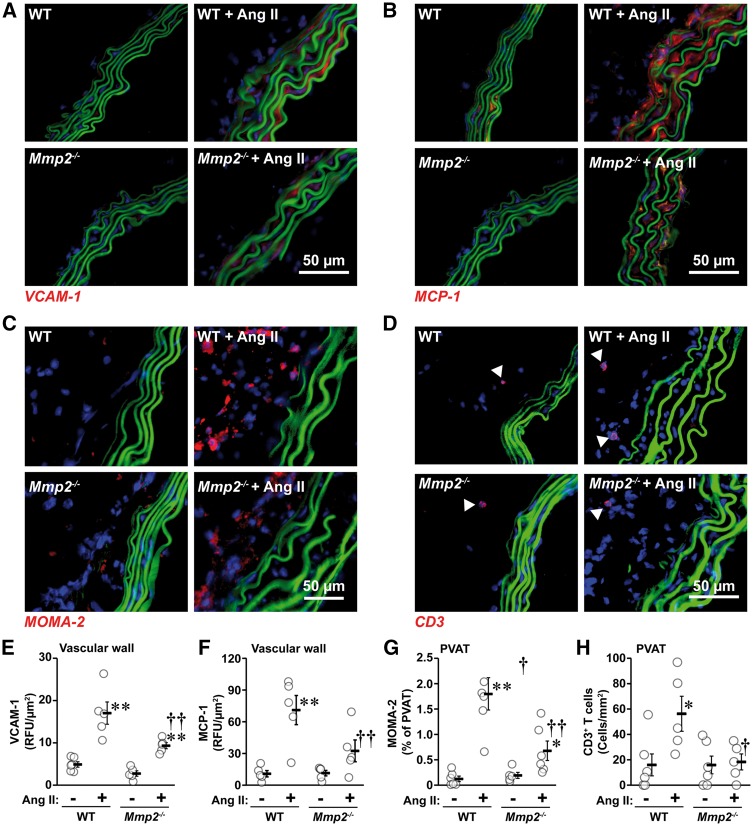
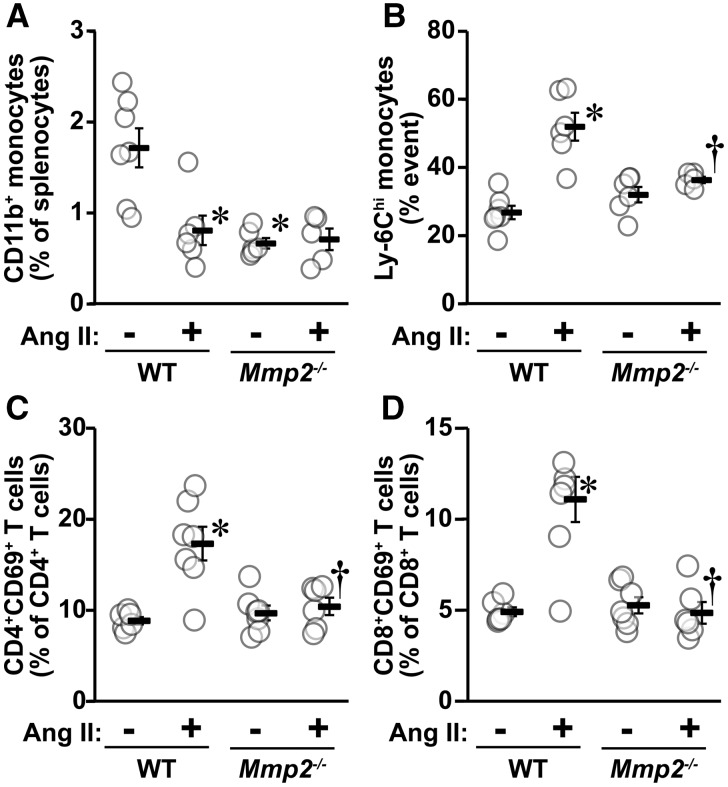
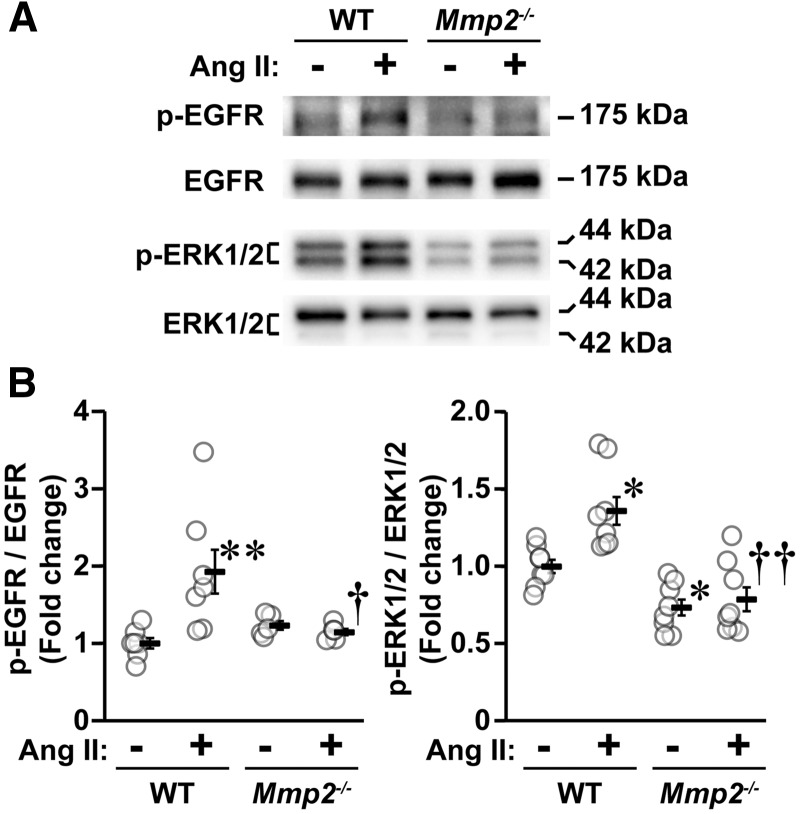
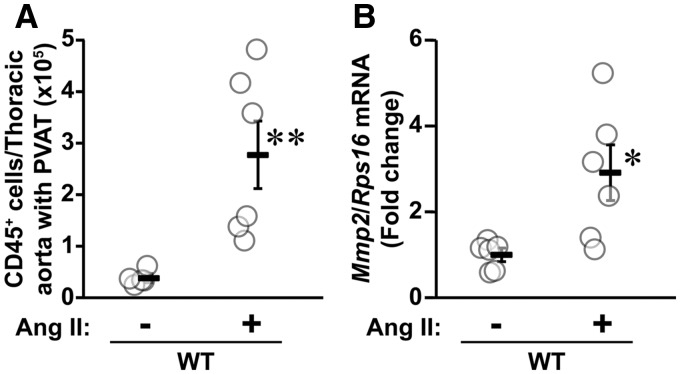
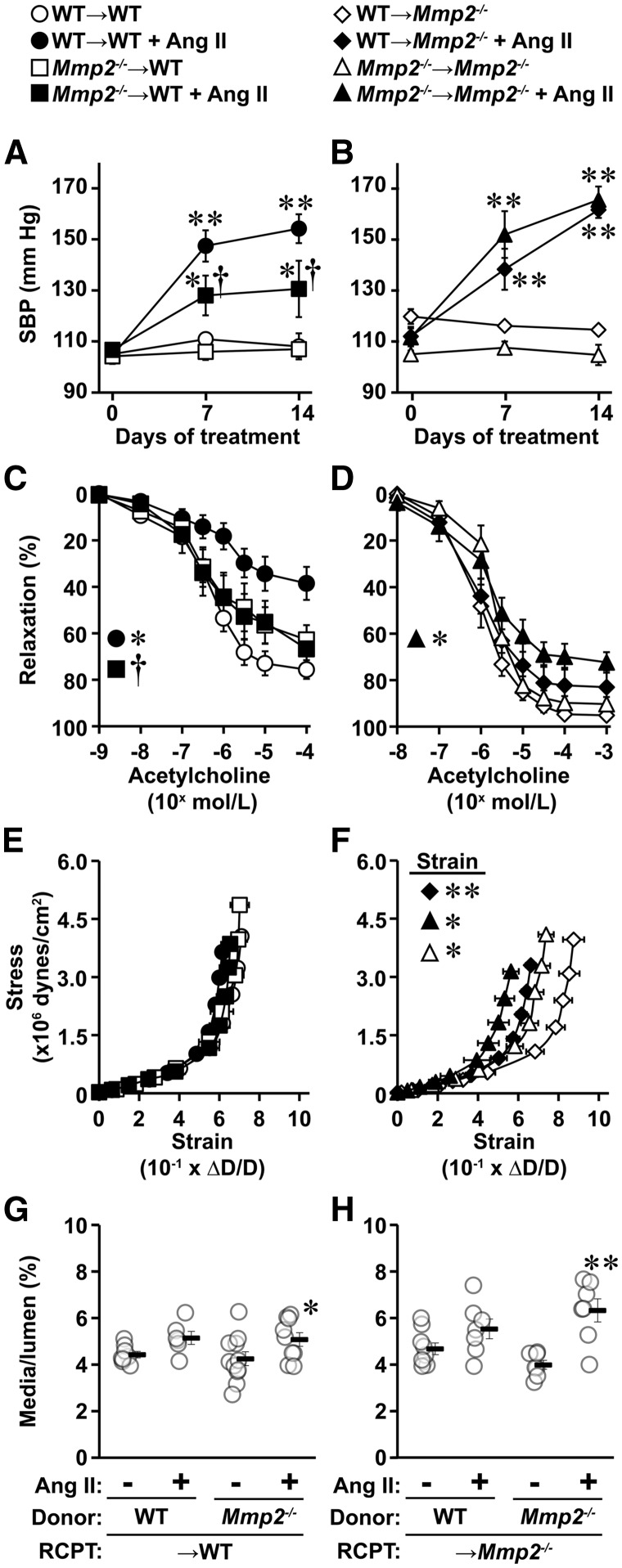
Methods and results: A fourteen-day Ang II infusion (1000 ng/kg/min, SC) increased systolic BP, decreased vasodilatory responses to acetylcholine, induced mesenteric artery (MA) hypertrophic remodelling, and enhanced MA stiffness in wild-type (WT) mice. Ang II enhanced aortic media and perivascular reactive oxygen species generation, aortic vascular cell adhesion molecule-1 and monocyte chemotactic protein-1 expression, perivascular monocyte/macrophage and T cell infiltration, and the fraction of spleen activated CD4+CD69+ and CD8+CD69+ T cells, and Ly-6Chi monocytes. Study of intracellular signalling showed that Ang II increased phosphorylation of epidermal growth factor receptor and extracellular-signal-regulated kinase 1/2 in vascular smooth muscle cells isolated from WT mice. All these effects were reduced or prevented by Mmp2 knockout, except for systolic BP elevation. Ang II increased Mmp2 expression in immune cells infiltrating the aorta and perivascular fat. Bone marrow (BM) transplantation experiments revealed that in absence of MMP2 in immune cells, Ang II-induced BP elevation was decreased, and that when MMP2 was deficient in either immune or vascular cells, Ang II-induced endothelial dysfunction was blunted.
Conclusions: Mmp2 knockout impaired Ang II-induced vascular injury but not BP elevation. BM transplantation revealed a role for immune cells in Ang II-induced BP elevation, and for both vascular and immune cell MMP2 in Ang II-induced endothelial dysfunction.
Keywords: Blood pressure; Bone marrow transplantation; Hypertension; MMP2; Vascular injury.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author 2017. For permissions, please email: journals.permissions@oup.com.
Figures
Comment in
-
Beyond the matrix: MMP2 as critical regulator of inflammation-mediated vascular dysfunction.Cardiovasc Res. 2017 Dec 1;113(14):1705-1707. doi: 10.1093/cvr/cvx202. Cardiovasc Res. 2017. PMID: 29040383 No abstract available.
References
-
- Schiffrin EL. Vascular remodeling in hypertension: mechanisms and treatment. Hypertension 2012;59:367–374. - PubMed
-
- Paradis P, Schiffrin EL, Renin–angiotensin–aldosterone system and pathobiology of hypertension. In DeMello WC, Frohlich ED (eds). Renin Angiotensin System and Cardiovascular Disease. New York, N.Y: Humana Press, 2009. pp. 35–58.
-
- Mian MO, Paradis P, Schiffrin EL.. Innate immunity in hypertension. Curr Hypertens Rep 2014;16:413.. - PubMed
-
- Fontana V, Silva PS, Gerlach RF, Tanus-Santos JE.. Circulating matrix metalloproteinases and their inhibitors in hypertension. Clin Chim Acta 2012;413:656–662. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
