

Triggered intracellular calcium waves in dog and human left atrial myocytes from normal and failing hearts
- PMID: 29016724
- PMCID: PMC5852523
- DOI: 10.1093/cvr/cvx167
Triggered intracellular calcium waves in dog and human left atrial myocytes from normal and failing hearts
Abstract
Aims: Abnormal intracellular Ca2+ cycling contributes to triggered activity and arrhythmias in the heart. We investigated the properties and underlying mechanisms for systolic triggered Ca2+ waves in left atria from normal and failing dog hearts.
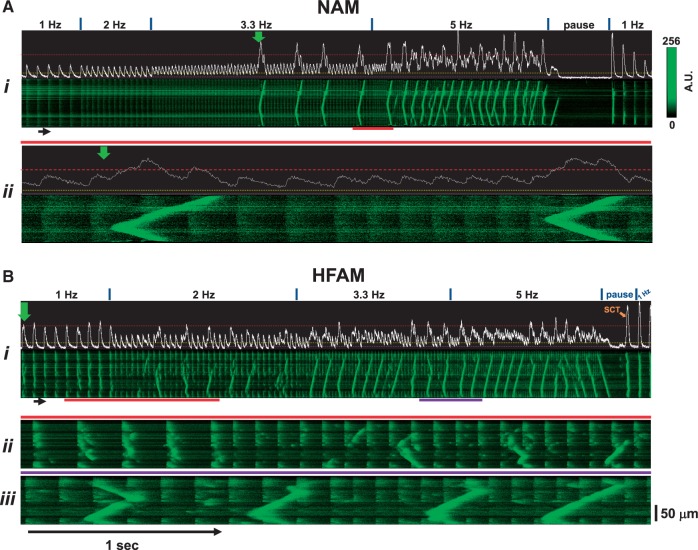
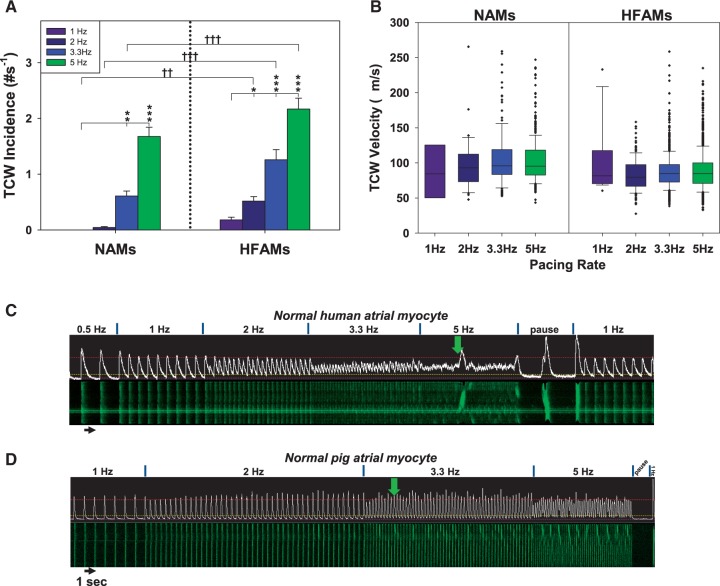
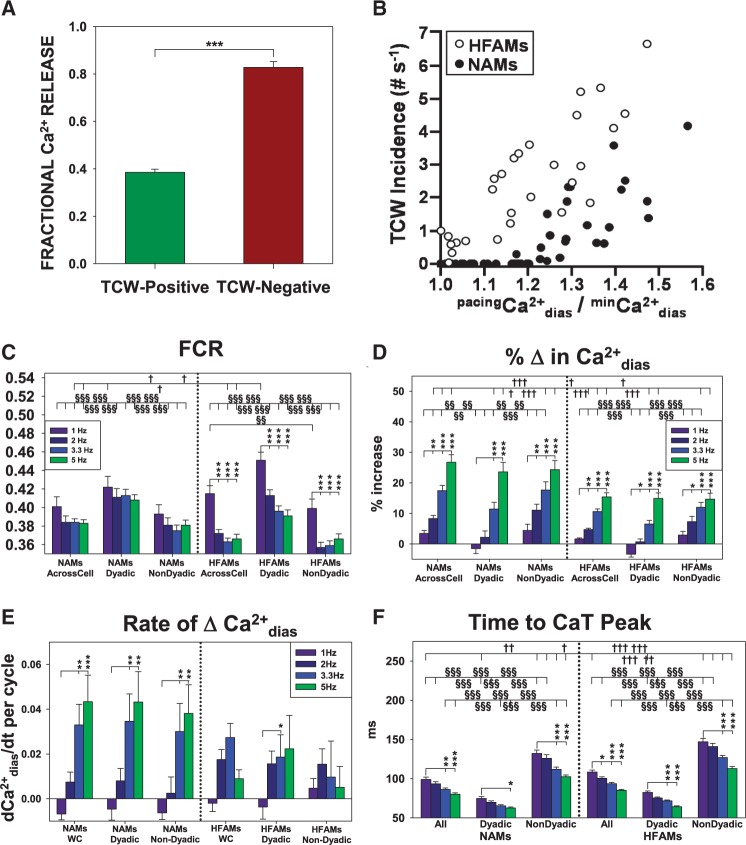
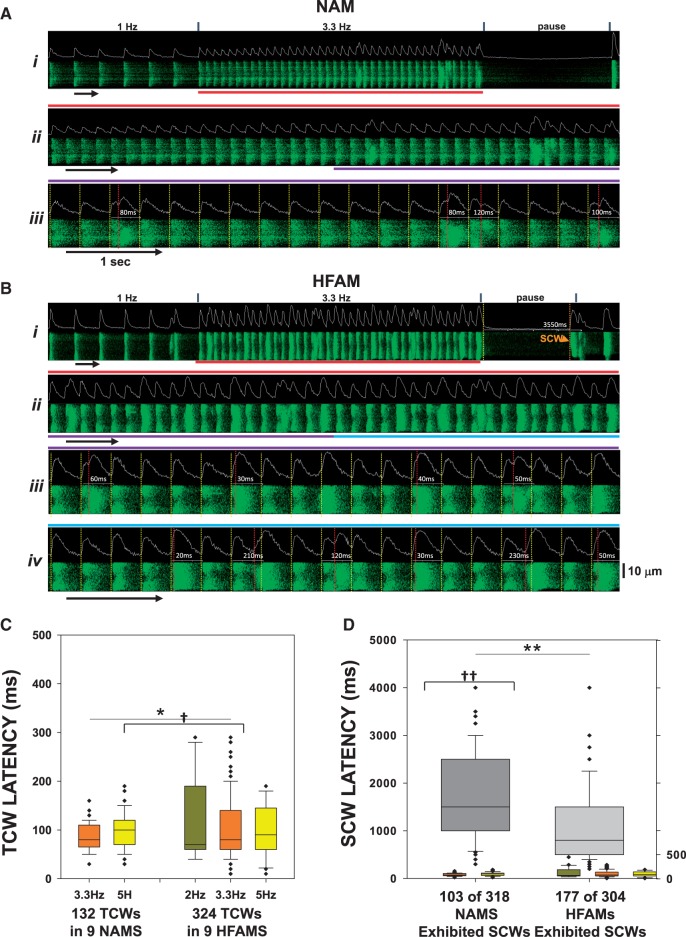
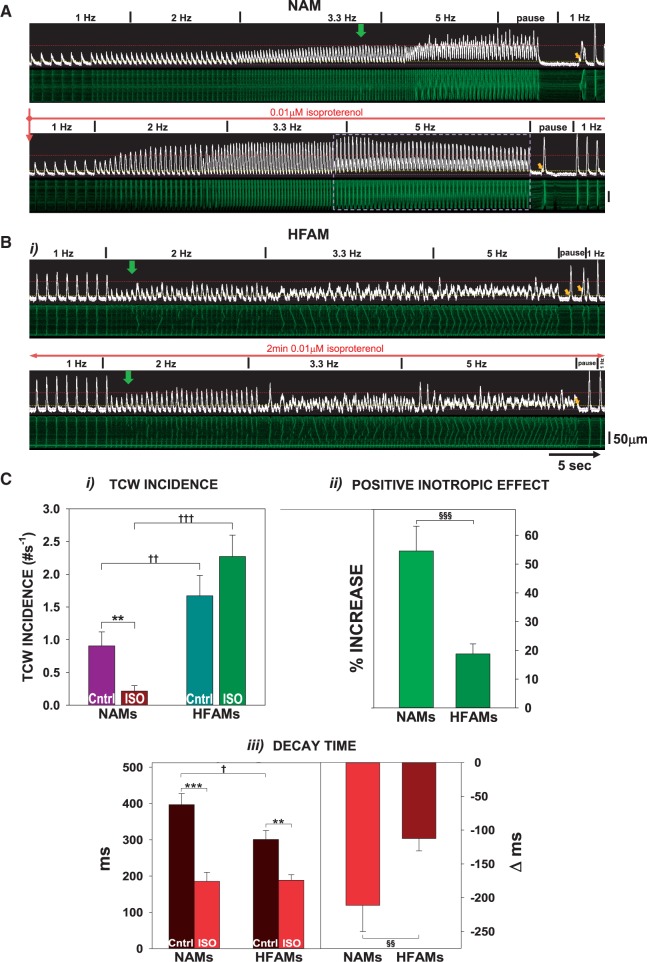
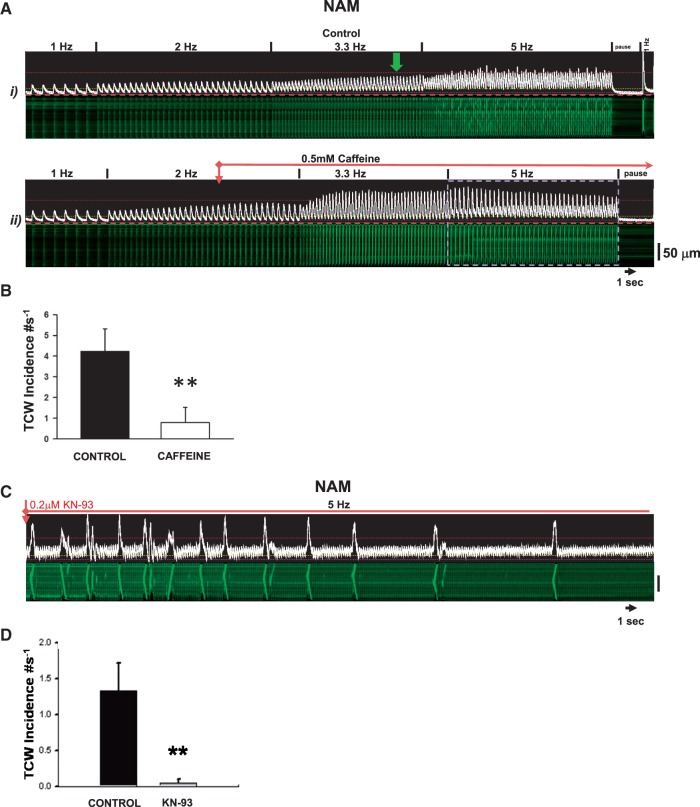
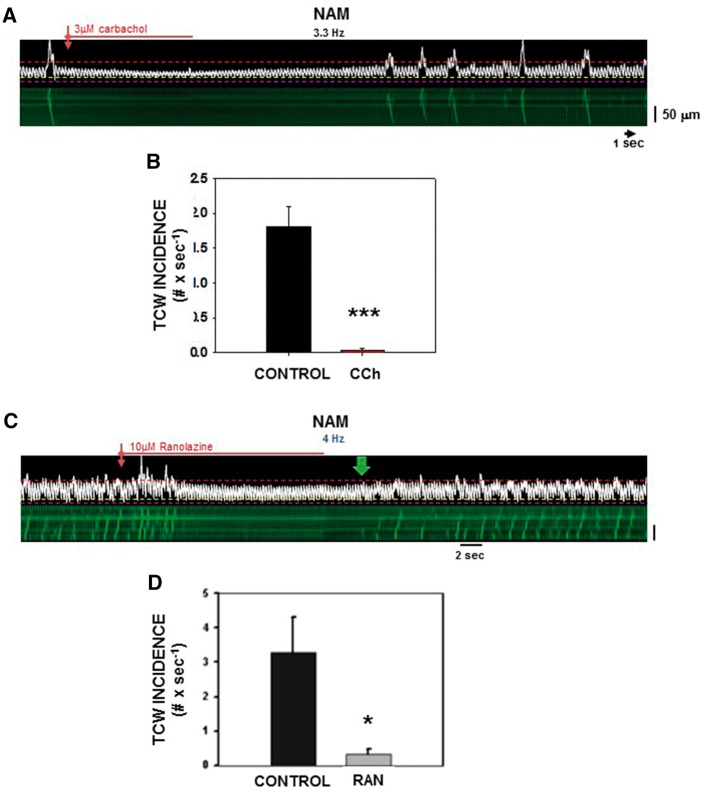
Methods and results: Intracellular Ca2+ cycling was studied using confocal microscopy during rapid pacing of atrial myocytes (36 °C) isolated from normal and failing canine hearts (ventricular tachypacing model). In normal atrial myocytes (NAMs), Ca2+ waves developed during rapid pacing at rates ≥ 3.3 Hz and immediately disappeared upon cessation of pacing despite high sarcoplasmic reticulum (SR) load. In heart failure atrial myocytes (HFAMs), triggered Ca2+ waves (TCWs) developed at a higher incidence at slower rates. Because of their timing, TCW development relies upon action potential (AP)-evoked Ca2+ entry. The distribution of Ca2+ wave latencies indicated two populations of waves, with early events representing TCWs and late events representing conventional spontaneous Ca2+ waves. Latency analysis also demonstrated that TCWs arise after junctional Ca2+ release has occurred and spread to non-junctional (cell core) SR. TCWs also occurred in intact dog atrium and in myocytes from humans and pigs. β-adrenergic stimulation increased Ca2+ release and abolished TCWs in NAMs but was ineffective in HFAMs making this a potentially effective adaptive mechanism in normals but potentially arrhythmogenic in HF. Block of Ca-calmodulin kinase II also abolished TCWs, suggesting a role in TCW formation. Pharmacological manoeuvres that increased Ca2+ release suppressed TCWs as did interventions that decreased Ca2+ release but these also severely reduced excitation-contraction coupling.
Conclusion: TCWs develop during the atrial AP and thus could affect AP duration, producing repolarization gradients and creating a substrate for reentry, particularly in HF where they develop at slower rates and a higher incidence. TCWs may represent a mechanism for the initiation of atrial fibrillation particularly in HF.
Keywords: Atrial fibrillation; Atrium; Heart failure; Ca2+ waves.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author 2017. For permissions please email: journals.permissions@oup.com.
Figures
References
-
- Aistrup GL, Balke CW, Wasserstrom JA.. Arrhythmia triggers in heart failure: the smoking gun of [Ca2+]i dysregulation. Heart Rhythm 2011;8:1804–1808. - PubMed
-
- Schotten U, Verheule S, Kirchhof P, Goette A.. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev 2011;91:265–325. - PubMed
-
- Greiser M, Kerfant BG, Williams GS, Voigt N, Harks E, Dibb KM, Giese A, Meszaros J, Verheule S, Ravens U, Allessie MA, Gammie JS, van der Velden J, Lederer WJ, Dobrev D, Schotten U.. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J Clin Invest 2014;124:4759–4772. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
