

DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway
- PMID: 29017571
- PMCID: PMC5635482
- DOI: 10.1186/s12964-017-0195-9
DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway
Abstract
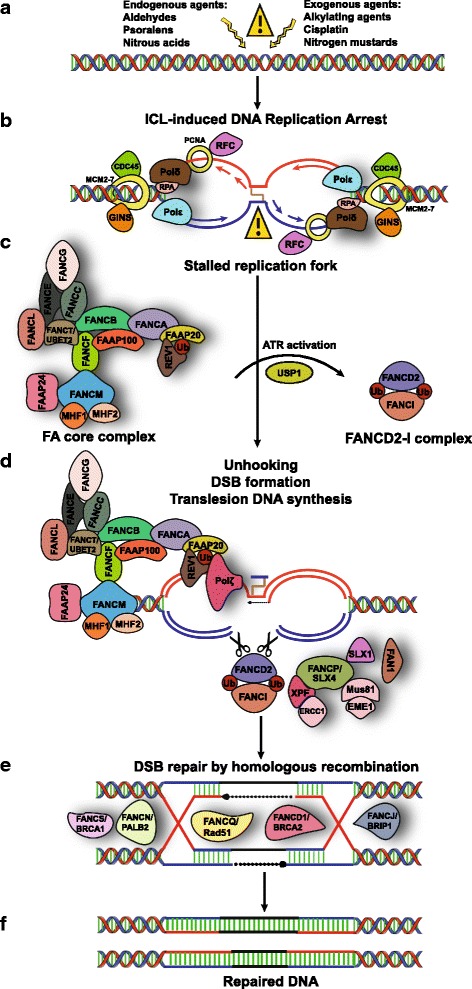
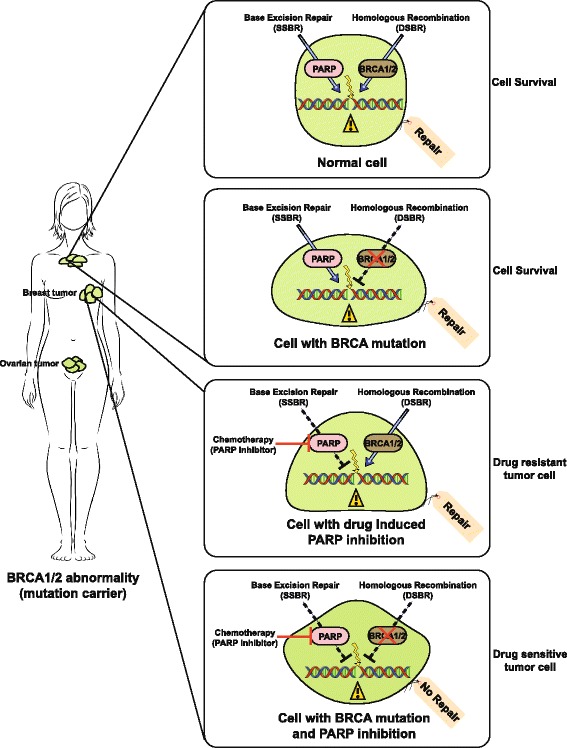
Fanconi Anemia (FA) is a rare, inherited genomic instability disorder, caused by mutations in genes involved in the repair of interstrand DNA crosslinks (ICLs). The FA signaling network contains a unique nuclear protein complex that mediates the monoubiquitylation of the FANCD2 and FANCI heterodimer, and coordinates activities of the downstream DNA repair pathway including nucleotide excision repair, translesion synthesis, and homologous recombination. FA proteins act at different steps of ICL repair in sensing, recognition and processing of DNA lesions. The multi-protein network is tightly regulated by complex mechanisms, such as ubiquitination, phosphorylation, and degradation signals that are critical for the maintenance of genome integrity and suppressing tumorigenesis. Here, we discuss recent advances in our understanding of how the FA proteins participate in ICL repair and regulation of the FA signaling network that assures the safeguard of the genome. We further discuss the potential application of designing small molecule inhibitors that inhibit the FA pathway and are synthetic lethal with DNA repair enzymes that can be used for cancer therapeutics.
Keywords: Cancer therapeutics; Combination Therapy Genomic instability; DNA damage response; DNA repair; Fanconi Anemia (FA) signaling network; Homologous recombination; Interstrand crosslink (ICL); Synthetic lethality; Translesion synthesis.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
