

doi: 10.1038/nbt.3956.
iML1515, a knowledgebase that computes Escherichia coli traits
Affiliations
- PMID: 29020004
- PMCID: PMC6521705
- DOI: 10.1038/nbt.3956
Item in Clipboard
iML1515, a knowledgebase that computes Escherichia coli traits
Nat Biotechnol.
.
No abstract available
Figures

iML1515 genome-scale reconstruction. (a) The iML1515 model contains 1,515 open reading frames that encode enzymes that catalyze 2,719 reactions involving 1,192 unique metabolites. It also includes 1,515 protein structures. All reconstruction content is linked to external databases, including KEGG, PDB, and CHEBI. iML1515 is capable of performing flux-balance analysis to integrate and interpret a variety of emerging data types including linking mutations identified from resequencing and/or transcriptomics data to fluxomics. (b) All reactions are linked to encoding gene(s) and protein. Connection to PDB structures and homology models form a domain-gene-protein-reaction relationship (dGPR) (Supplementary Data set 7). (c) Clustering of domain architecture and metabolite usage provides tools to explore enzyme promiscuity and metabolism,. The domain-connectivity network can be visualized using Cytoscape and is supplied as a network file (Supplementary Data set 8). The acetyltransferase domain in c highlights a specific example of domain connectivity. The acyltransferase domain (d1iuqa_) is present in three genes (b3018, b1054, and b2378). The encoded proteins catalyze different but related reactions in glycerophospholipid metabolism and endotoxin synthesis. All reactions are ACP-dependent acyltransferases. (d) A database consisting of 333 normalized transcriptomics data sets was contextualized using the GPRs of iML1515. Relative expression for all three genes catalyzing the ASPK reaction are plotted across all experimental conditions, revealing condition-specific preferences for gene usage. The experimental conditions that favor a particular isozyme are listed. At the top of the panel two reactions (ASPK, HSDy) are shown with two isozymes that can catalyze these reactions (ThrA, MetL). The third isozyme (LysC) can only catalyze APSK. ASPK and HSDy activity must be present to synthesize L-threonine (thr-L), L-methionine (met-L), L-isoleucine (ile-L), biotin (btn), and S-adenosyl-L-methionine (amet). Only ASPK activity is needed to synthesize murein derivatives and L-lysine (lys-L) (further discussion can be found in Supplementary Fig. 8 and Supplementary Note 6).

Model validation. (a) The Colony-live platform was used to measure growth capabilities of 3,869 single-knockout mutant E. coli strains on minimal media with 16 different carbon sources, forming a total of 62,272 measured phenotypes. Colony-live provides specific values for lag-time (LTG), maximum growth rate (MGR), and growth saturation point (GSP) for each gene knockout and condition (presented in Supplementary Data set 11). (b) Subset of knockout data highlighting growth rates for gene knockouts in the tricarboxylic acid (citric acid) cycle. (c) The iML1515 reconstruction is 93.4% accurate in predicting the effect of gene knockouts, an increase in accuracy of 3.7% over the 89.8% accuracy of the iJO1366 E. coli metabolic reconstruction.
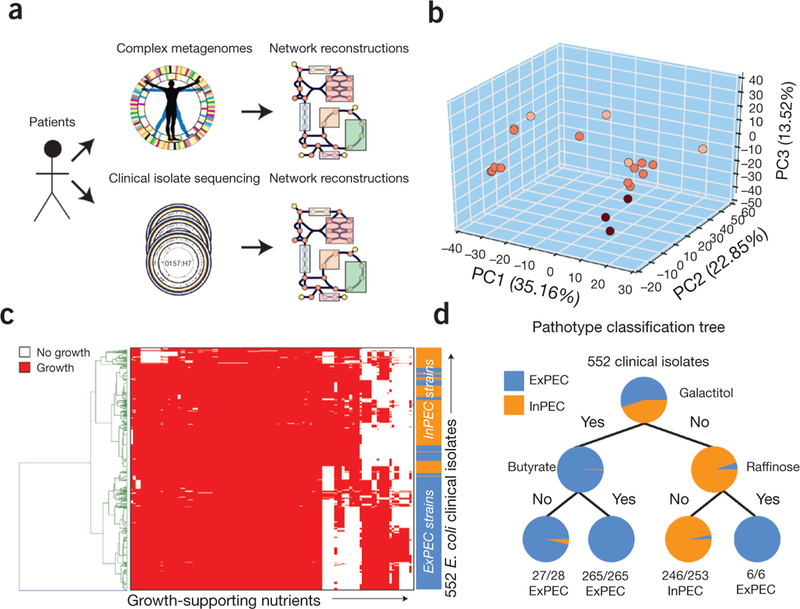
Application of iML1515 for clinical isolates and metagenomes. (a) iML1515 can be used to rapidly construct strain-specific models of metabolism from sequenced clinical isolates and complex metagenomes. Genes that are part of the iML1515 model are identified and extracted for comparison across each of the metagenomics samples. (b) Sample-specific models of E. coli metabolism were constructed for 22 metagenomic samples by evaluating shared content from iML1515. Metabolite synthesis capabilities and yields were calculated for each model and evaluated using PCA to illustrate a separation in sample-specific metabolite synthesis capabilities. Points are colored based on model-predicted max autoinducer-2 yield. (c) Strain-specific models were constructed for 552 E. coli clinical isolates from two recent studies,. Models were used to predict the ability to grow on over 300 different carbon, nitrogen, phosphorous, and sulfur sources. A heatmap of model-predicted catabolic capabilities for clinical isolates is shown. (d) Machine learning methods, such as a decision tree, can be applied to model predictions. For example, model-predicted catabolic capabilities can be used to classify clinical isolates between extra-intestinal pathotypes (ExPEC: isolated from blood or urine) and from intestinal pathotypes (isolated from feces) based solely on the model-predicted ability to catabolize three substrates (galactitol, butyrate, and raffinose).

Comparison of iML1515 to sequence variations in 1,122 clinical Isolates of E. coli. (a) Counts for each E. coli K-12 MG1655 gene in 1,122 sequenced strains of E. coli are shown. (b) Histidine pathways showed high levels of amino acid differences among genes involved. The pie chart represents the percentage of strains that contain unique hisDalleles. The hisD116 allele of E. coli K-12 MG1655 is present in only 19 (1.7%) of the sequenced strains. (c) Structural biology methods can reveal the effect of mutations. (d) 976 genes with metabolic functions are conserved across 99% of E. coli strains and this core set also has mutations. The bar chart shows the average number of amino acid mutations in these core genes for 1,122 strains of E. coli. (e) A histogram showing how many genes have a certain number of average mutations. (f) Amino acid mutations are compared for major metabolic pathways (e.g., histidine synthesis) using structural biology methods. (g) Amino acid changes can be compared per protein domain. For example, genes that encode the aldehyde dehydrogenase (ALDH-like) domain present in hisD have, on average, more genetic mutations across all 1,122 strains of E. coli than genes encoding other domains.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
