

Activation of Autophagy Ameliorates Cardiomyopathy in Mybpc3-Targeted Knockin Mice
- PMID: 29021349
- PMCID: PMC5679453
- DOI: 10.1161/CIRCHEARTFAILURE.117.004140
Activation of Autophagy Ameliorates Cardiomyopathy in Mybpc3-Targeted Knockin Mice
Abstract
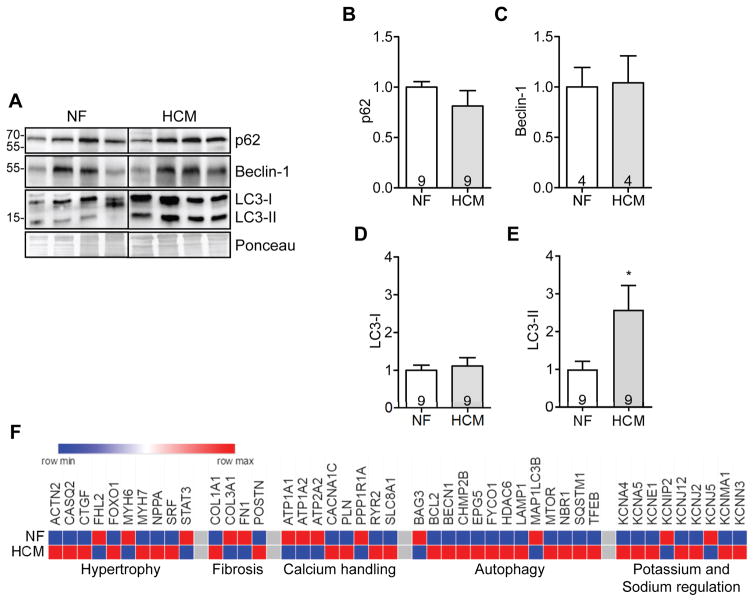
Background: Alterations in autophagy have been reported in hypertrophic cardiomyopathy (HCM) caused by Danon disease, Vici syndrome, or LEOPARD syndrome, but not in HCM caused by mutations in genes encoding sarcomeric proteins, which account for most of HCM cases. MYBPC3, encoding cMyBP-C (cardiac myosin-binding protein C), is the most frequently mutated HCM gene.
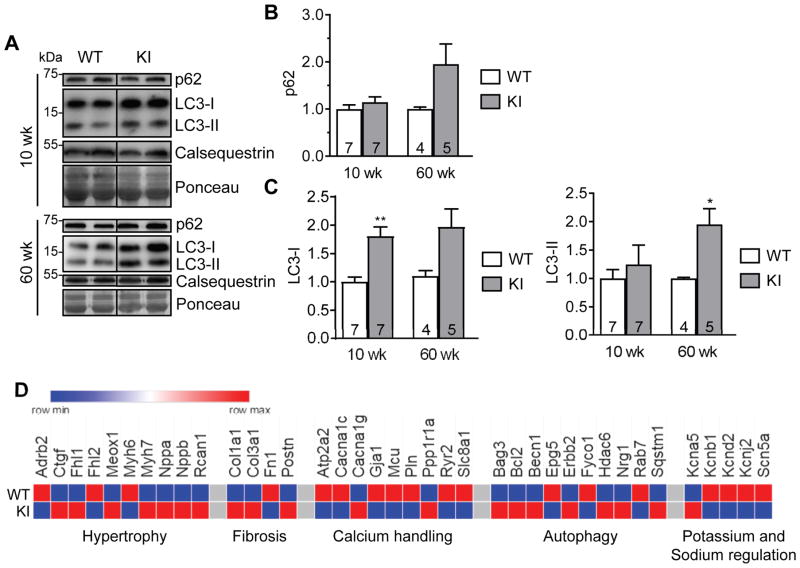
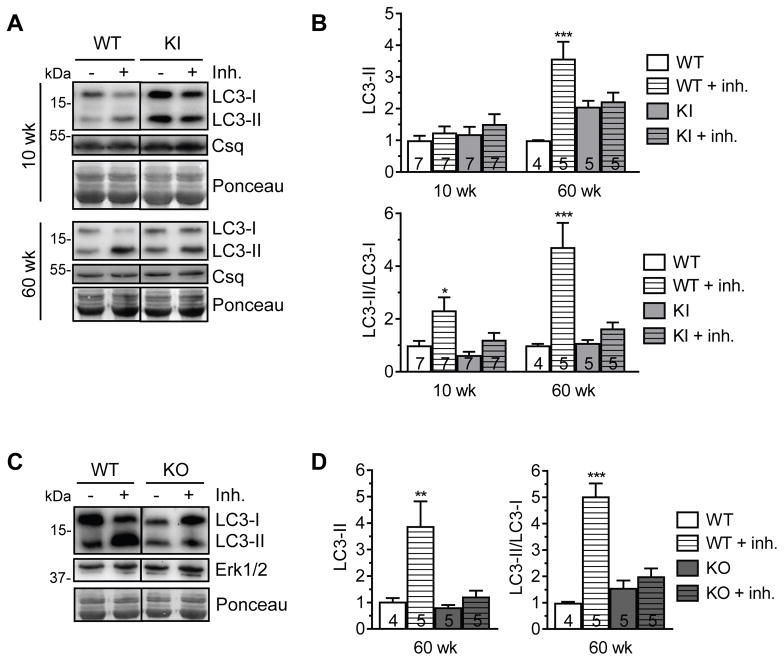
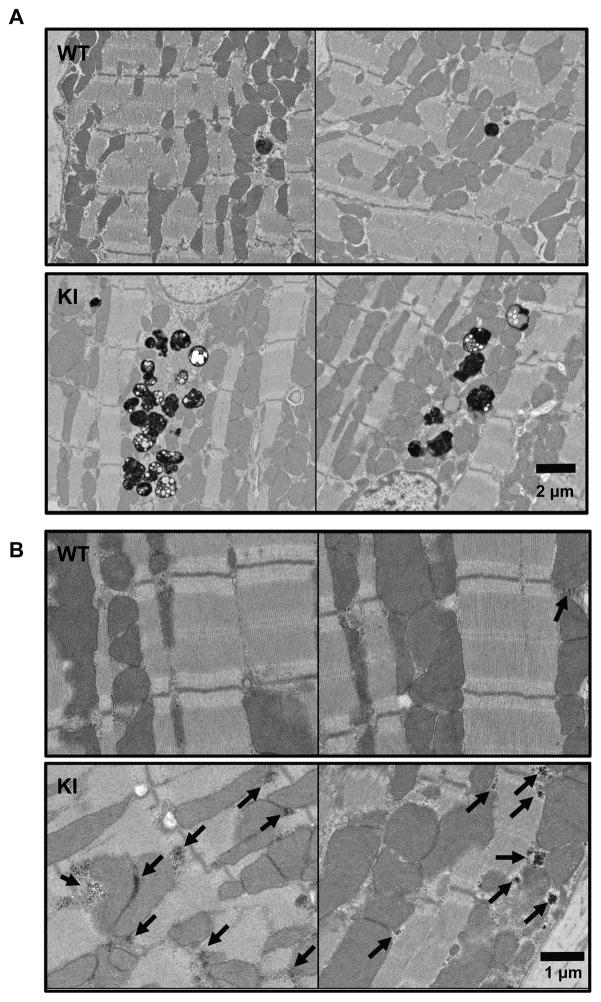
Methods and results: We evaluated autophagy in patients with HCM carrying MYBPC3 mutations and in a Mybpc3-targeted knockin HCM mouse model, as well as the effect of autophagy modulators on the development of cardiomyopathy in knockin mice. Microtubule-associated protein 1 light chain 3 (LC3)-II protein levels were higher in HCM septal myectomies than in nonfailing control hearts and in 60-week-old knockin than in wild-type mouse hearts. In contrast to wild-type, autophagic flux was blunted and associated with accumulation of residual bodies and glycogen in hearts of 60-week-old knockin mice. We found that Akt-mTORC1 (mammalian target of rapamycin complex 1) signaling was increased, and treatment with 2.24 mg/kg·d rapamycin or 40% caloric restriction for 9 weeks partially rescued cardiomyopathy or heart failure and restored autophagic flux in knockin mice.
Conclusions: Altogether, we found that (1) autophagy is altered in patients with HCM carrying MYBPC3 mutations, (2) autophagy is impaired in Mybpc3-targeted knockin mice, and (3) activation of autophagy ameliorated the cardiac disease phenotype in this mouse model. We propose that activation of autophagy might be an attractive option alone or in combination with another therapy to rescue HCM caused by MYBPC3 mutations.
Keywords: autophagy; caloric restriction; cardiomyopathy; hypertrophy; rapamycin.
© 2017 American Heart Association, Inc.
Conflict of interest statement
Figures
References
-
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–6. - PubMed
-
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435. - PubMed
-
- Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–57. - PubMed
-
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, Sue CM, Yamamoto A, Murakami N, Shanske S, Byrne E, Bonilla E, Nonaka I, DiMauro S, Hirano M. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–10. - PubMed
-
- Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S, Schliselfeld LH. Lysosomal glycogen storage disease with normal acid maltase. Neurology. 1981;31:51–7. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
