

HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients
- PMID: 29021520
- PMCID: PMC5636840
- DOI: 10.1038/s41467-017-00911-y
HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients
Abstract
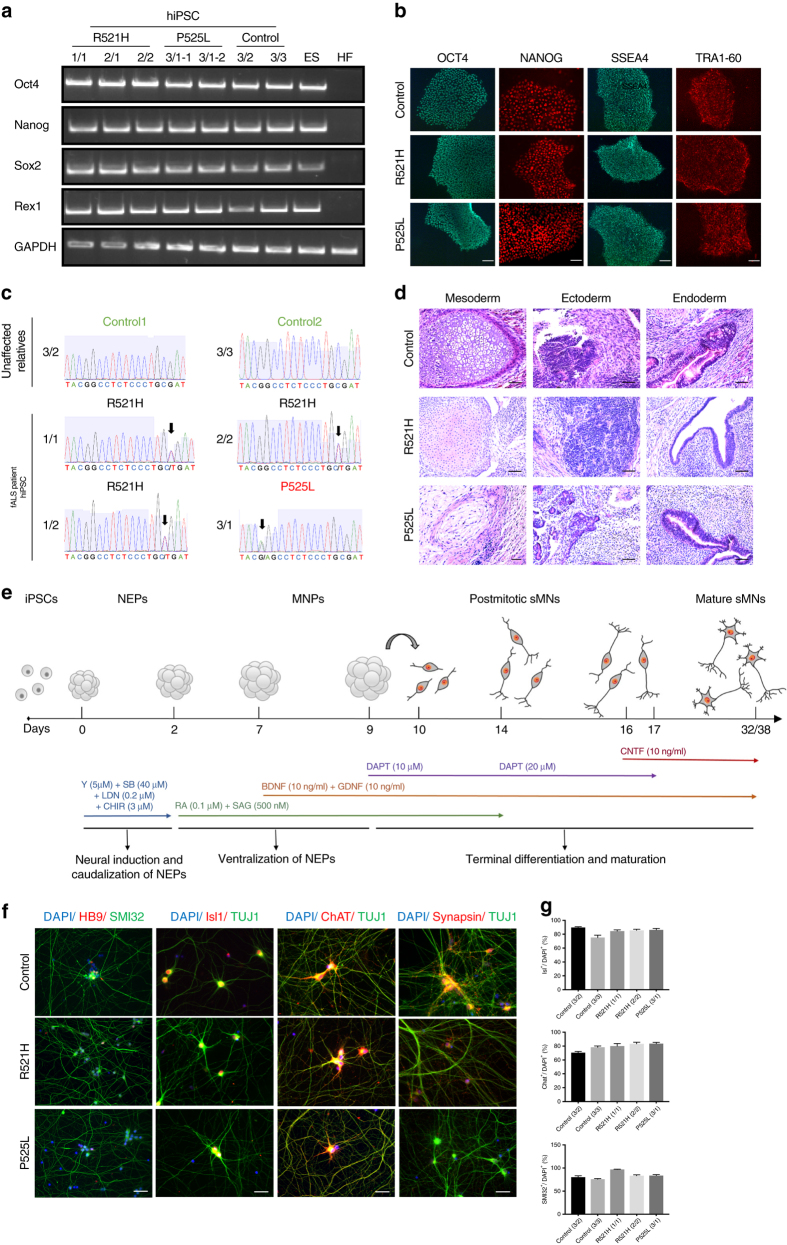
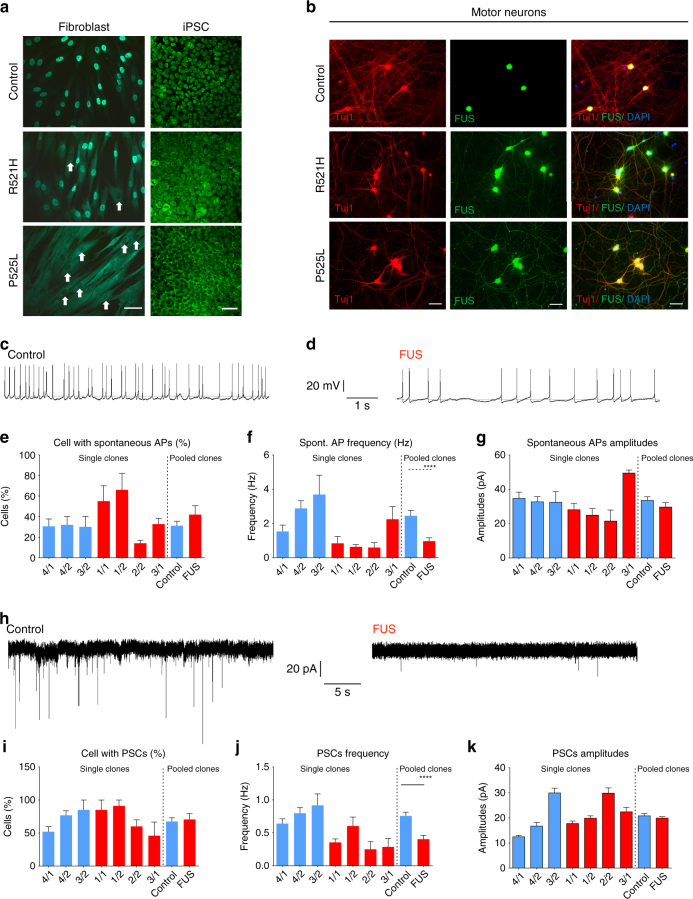
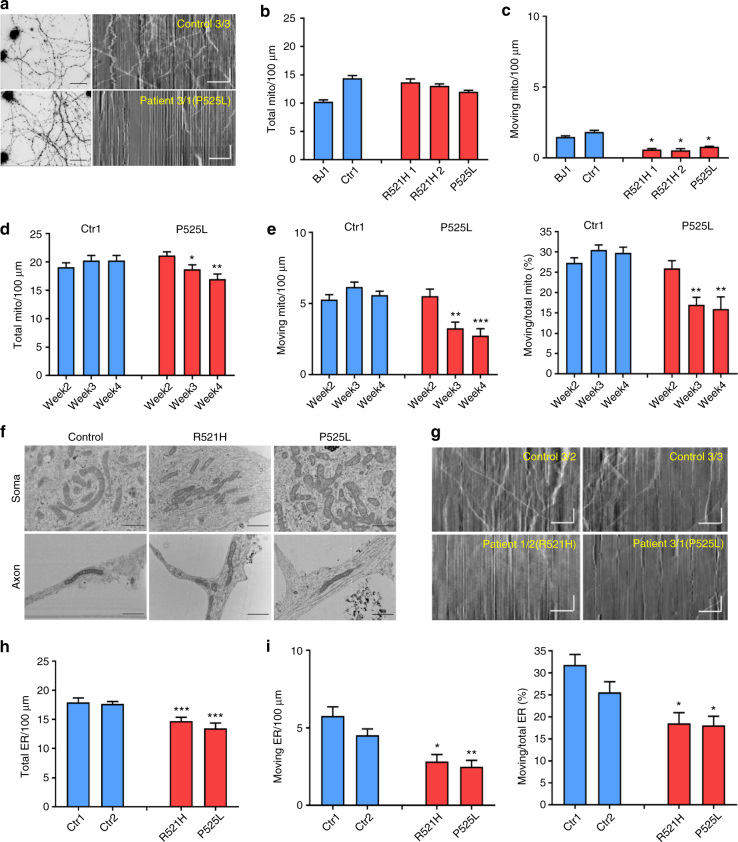
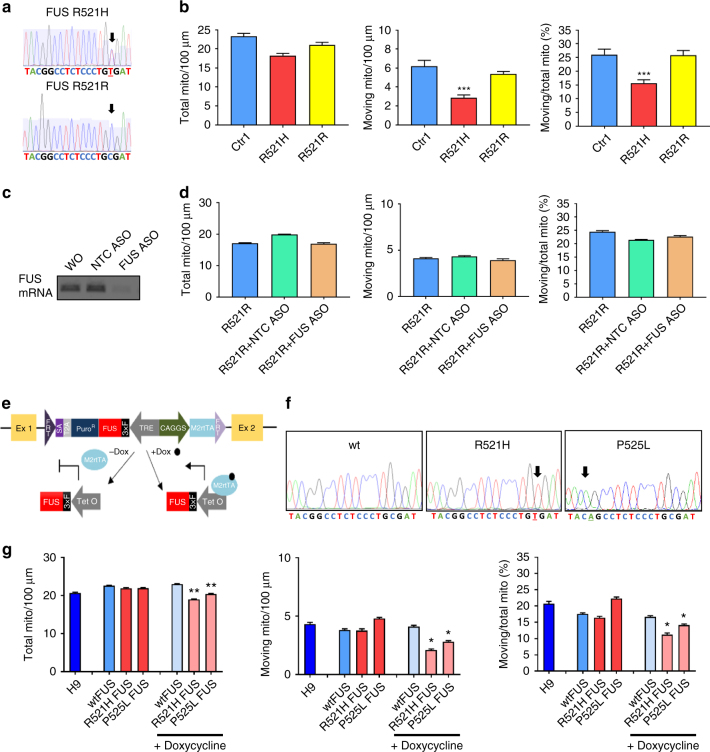
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disorder due to selective loss of motor neurons (MNs). Mutations in the fused in sarcoma (FUS) gene can cause both juvenile and late onset ALS. We generated and characterized induced pluripotent stem cells (iPSCs) from ALS patients with different FUS mutations, as well as from healthy controls. Patient-derived MNs show typical cytoplasmic FUS pathology, hypoexcitability, as well as progressive axonal transport defects. Axonal transport defects are rescued by CRISPR/Cas9-mediated genetic correction of the FUS mutation in patient-derived iPSCs. Moreover, these defects are reproduced by expressing mutant FUS in human embryonic stem cells (hESCs), whereas knockdown of endogenous FUS has no effect, confirming that these pathological changes are mutant FUS dependent. Pharmacological inhibition as well as genetic silencing of histone deacetylase 6 (HDAC6) increase α-tubulin acetylation, endoplasmic reticulum (ER)-mitochondrial overlay, and restore the axonal transport defects in patient-derived MNs.Amyotrophic lateral sclerosis (ALS) leads to selective loss of motor neurons. Using motor neurons derived from induced pluripotent stem cells from patients with ALS and FUS mutations, the authors demonstrate that axonal transport deficits that are observed in these cells can be rescued by HDAC6 inhibition.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
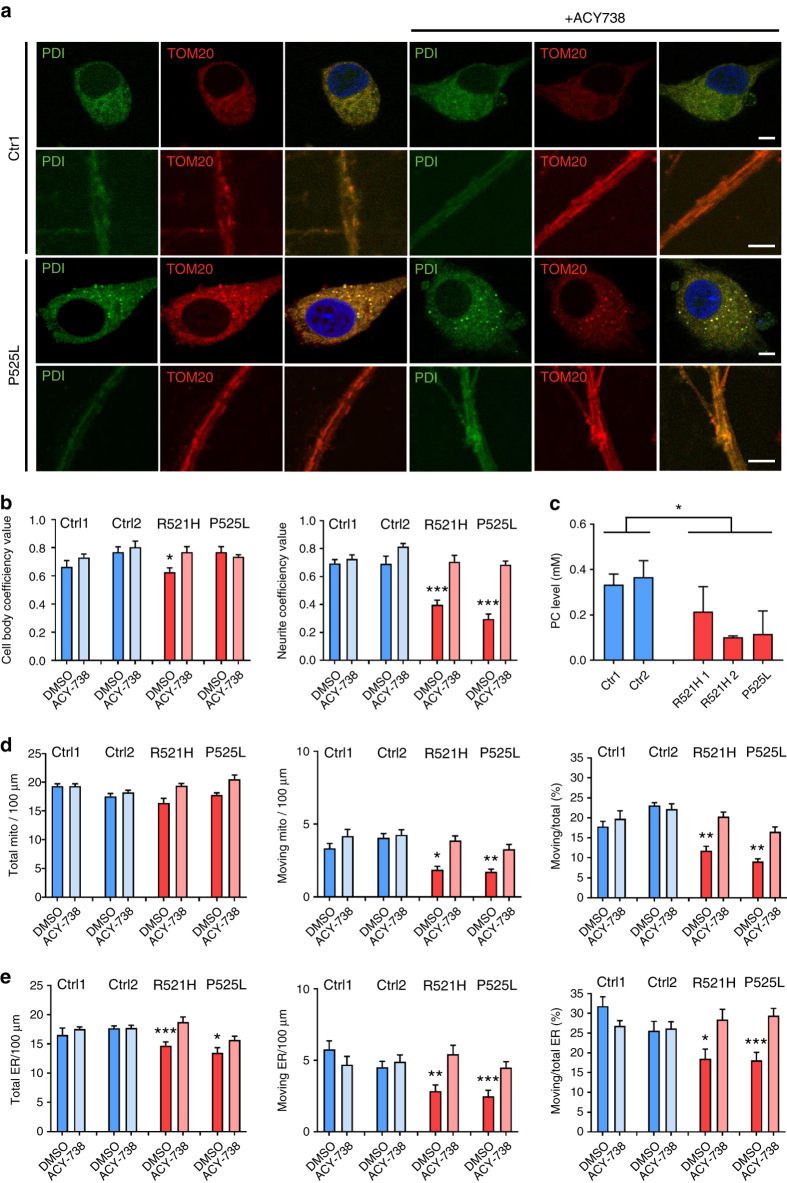
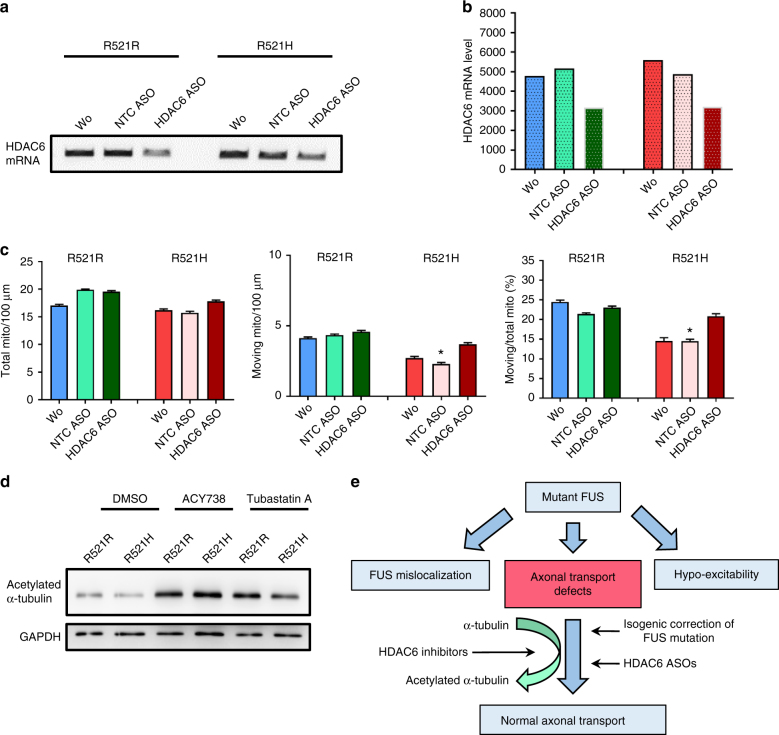
Comment in
-
Therapeutic potential of HDAC6 in amyotrophic lateral sclerosis.Cell Stress. 2017 Dec 19;2(1):14-16. doi: 10.15698/cst2018.01.120. Cell Stress. 2017. PMID: 31225447 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
