

Dysfunction of Membrane Trafficking Leads to Ischemia-Reperfusion Injury After Transient Cerebral Ischemia
- PMID: 29022237
- PMCID: PMC5895539
- DOI: 10.1007/s12975-017-0572-0
Dysfunction of Membrane Trafficking Leads to Ischemia-Reperfusion Injury After Transient Cerebral Ischemia
Abstract
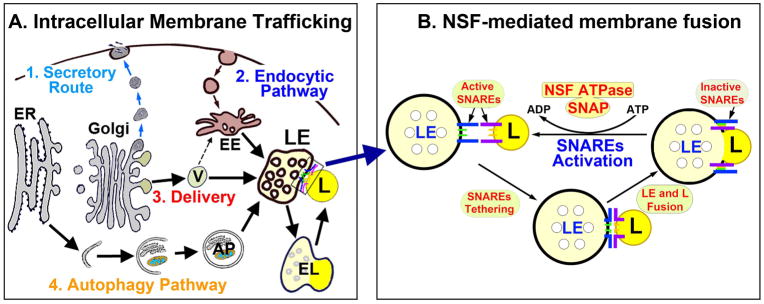
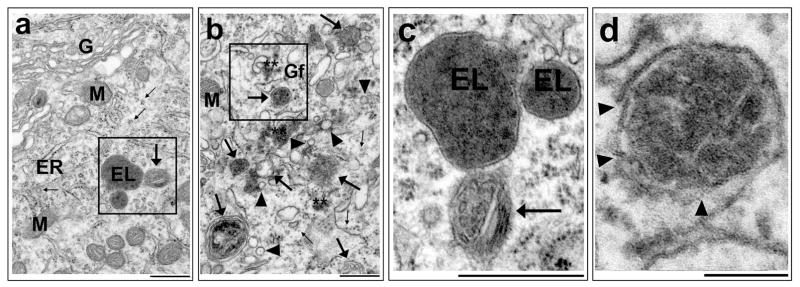
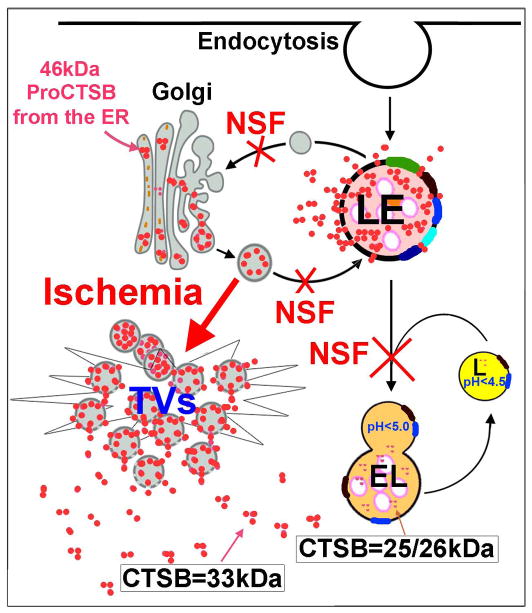
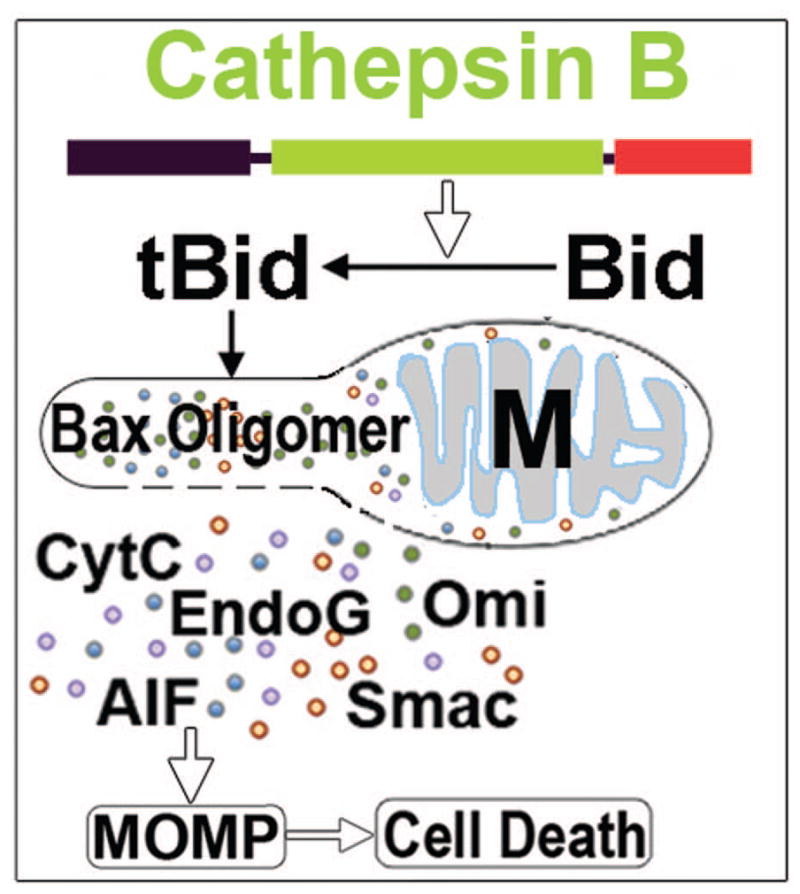
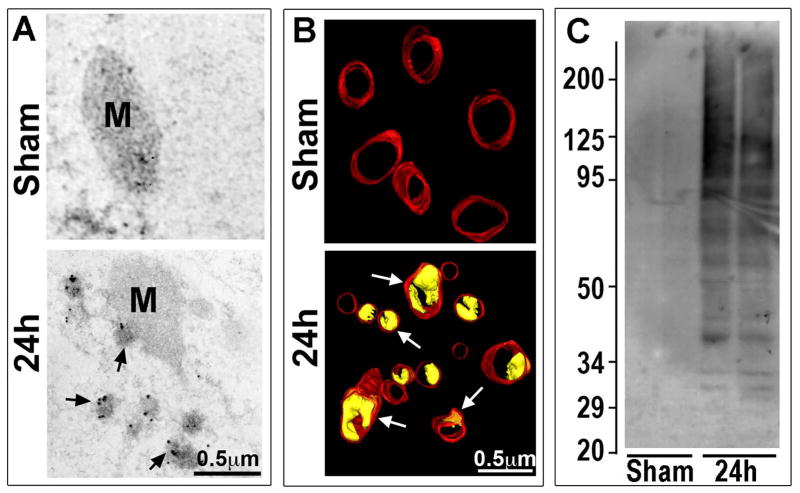
Neurons require an extraordinarily high level of membrane trafficking activities because of enriched axonal terminals and dendritic branches. For that reason, defects in the membrane trafficking pathway are a hallmark of most, and may be all, neurodegenerative disorders. A major cellular membrane trafficking pathway is the Golgi apparatus (Golgi hereafter)-late endosome-lysosome axis for supplying lysosomal enzymes. This pathway is regulated by N-ethylmaleimide-sensitive factor (NSF) ATPase. This review article is to discuss a novel hypothesis that brain ischemia inactivates NSF ATPase, resulting in a cascade of events of disruption of the Golgi-endosome-lysosome pathway, release of cathepsin B (CTSB), and induction of mitochondrial outer membrane permeabilization (MOMP) during the postischemic phase. This hypothesis is supported by recent studies demonstrating that NSF is trapped into inactive protein aggregates in neurons destined to die after brain ischemia. Consequently, Golgi, transport vesicles (TVs), and late endosomes (LEs) are accumulated and damaged, which is followed by CTSB release from these damaged structures. Moderate release of CTSB cleaves Bax-like BH3 protein (Bid) to become active truncated Bid (tBid). Active tBid is then translocated to the mitochondrial outer membrane, resulting in oligomerization of BCL2-associated X protein (Bax) forming the mitochondrial outer membrane pores, and releasing mitochondrial intramembranous proteins. Extensive CTSB release, however, can digest cellular proteins indiscriminately to induce cell death. Based on these new observations, we propose a novel hypothesis, i.e., brain ischemia leads to NSF inactivation, resulting in a massive buildup of damaged Golgi, TVs and LEs, fatal release of CTSB, induction of MOMP, and eventually brain ischemia-reperfusion injury.
Keywords: Brain ischemia-reperfusion injury; Golgi; Late endosome; Lysosome; Membrane trafficking; Mitochondrial outer membrane permeabilization; NSF, cathepsin B; Transport vesicle.
Figures

References
-
- Smith ML, Auer RN, Siesjo BK. The density and distribution of ischemic brain injury in the rat following 2–10 min of forebrain ischemia. Acta Neuropathol (Berl) 1984;64:319–332. - PubMed
-
- Taniguchi D, Baernstein A, Nichol G. Cardiac arrest: a public health perspective. Emerg Med Clin North Am. 2012 Feb;30(1):1–12. - PubMed
-
- Kirino T, Tamura A, Sano K. Delayed neuronal death in the rat hippocampus following transient forebrain ischemia. Acta Neuropathol. 1984;64:139–147. - PubMed
-
- Kirino T, Sano K. Fine structural nature of delayed neuronal death following ischemia in the gerbil hippocampus. Acta Neuropathol. 1984;62:209–218. - PubMed
-
- Lin B, Ginsberg MD, Busto R. Hyperglycemic exacerbation of neuronal damage following forebrain ischemia: microglial, astrocytic and endothelial alterations. Acta Neuropathol. 1998;96:610–620. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
