

The impact of rare variation on gene expression across tissues
- PMID: 29022581
- PMCID: PMC5877409
- DOI: 10.1038/nature24267
The impact of rare variation on gene expression across tissues
Abstract
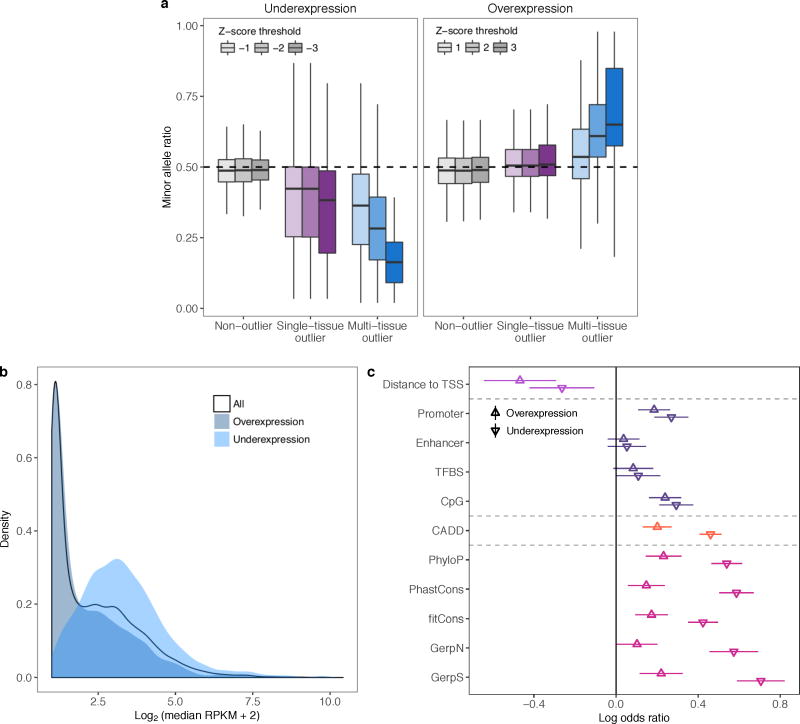
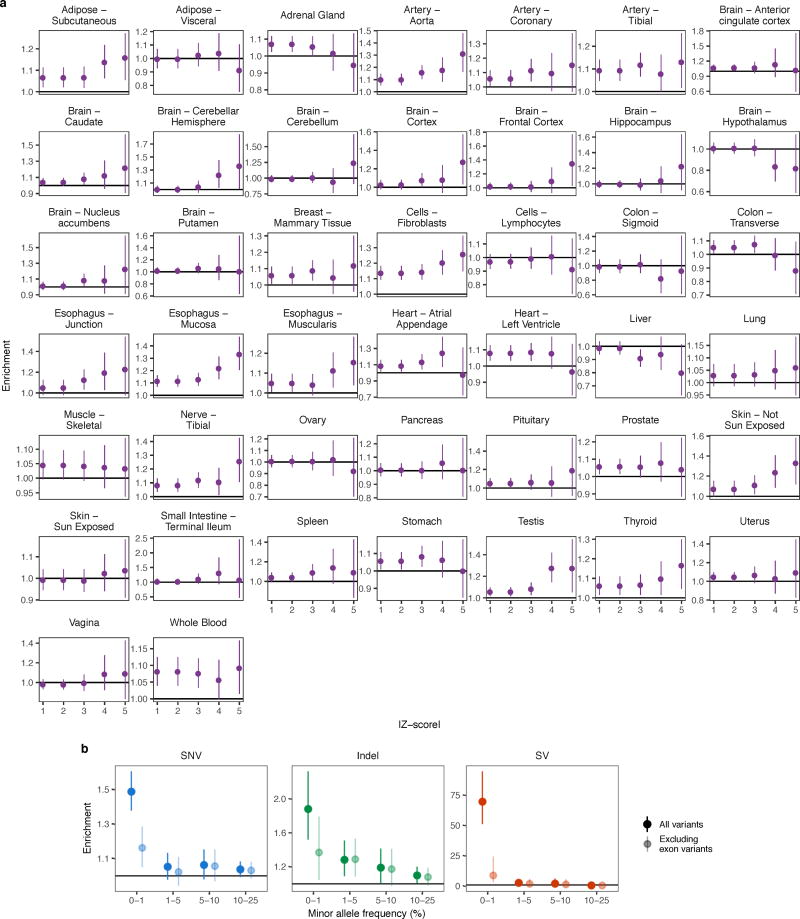
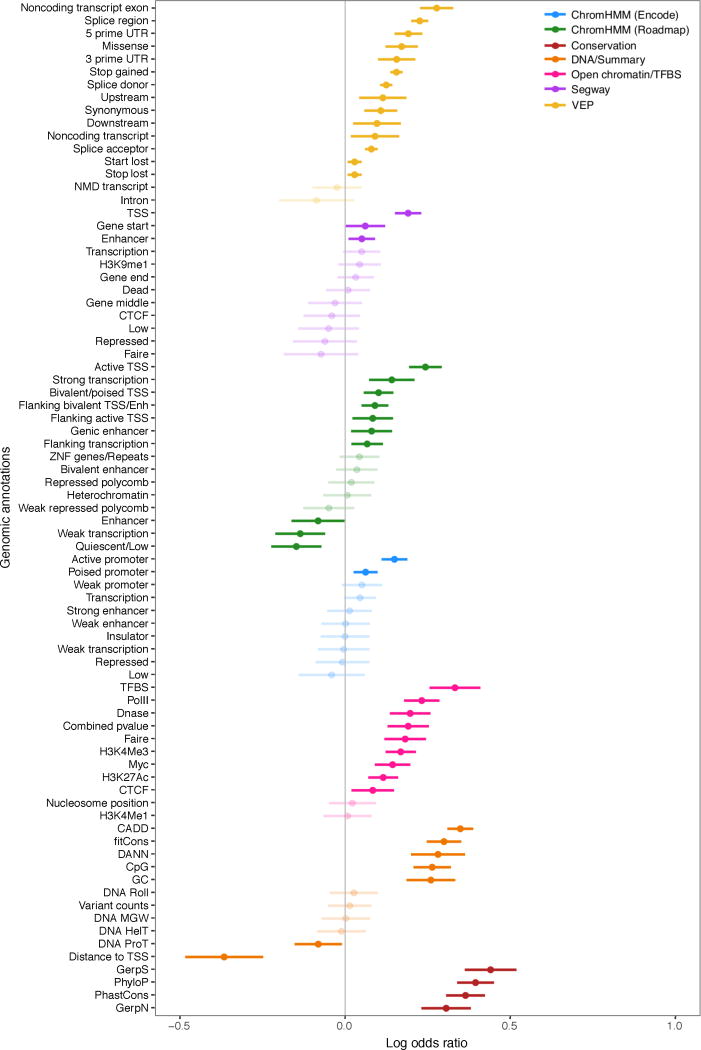
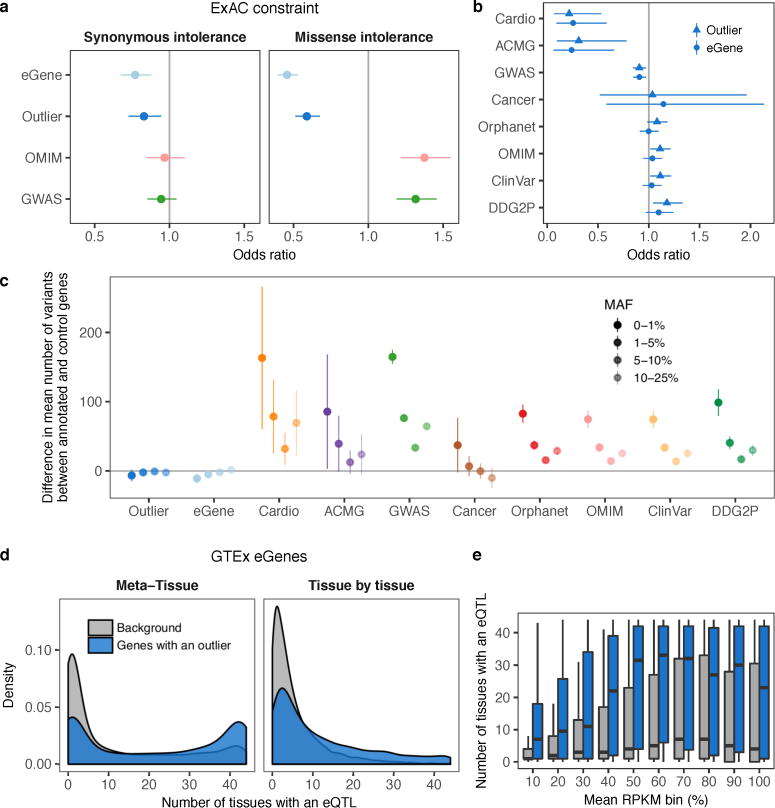
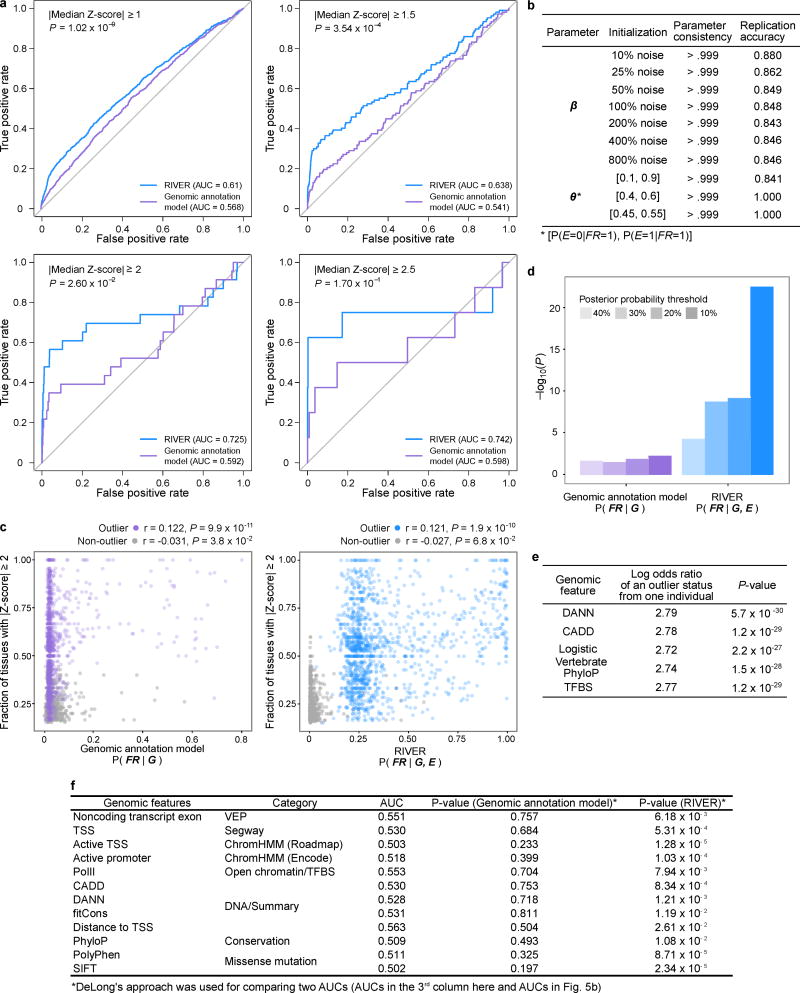
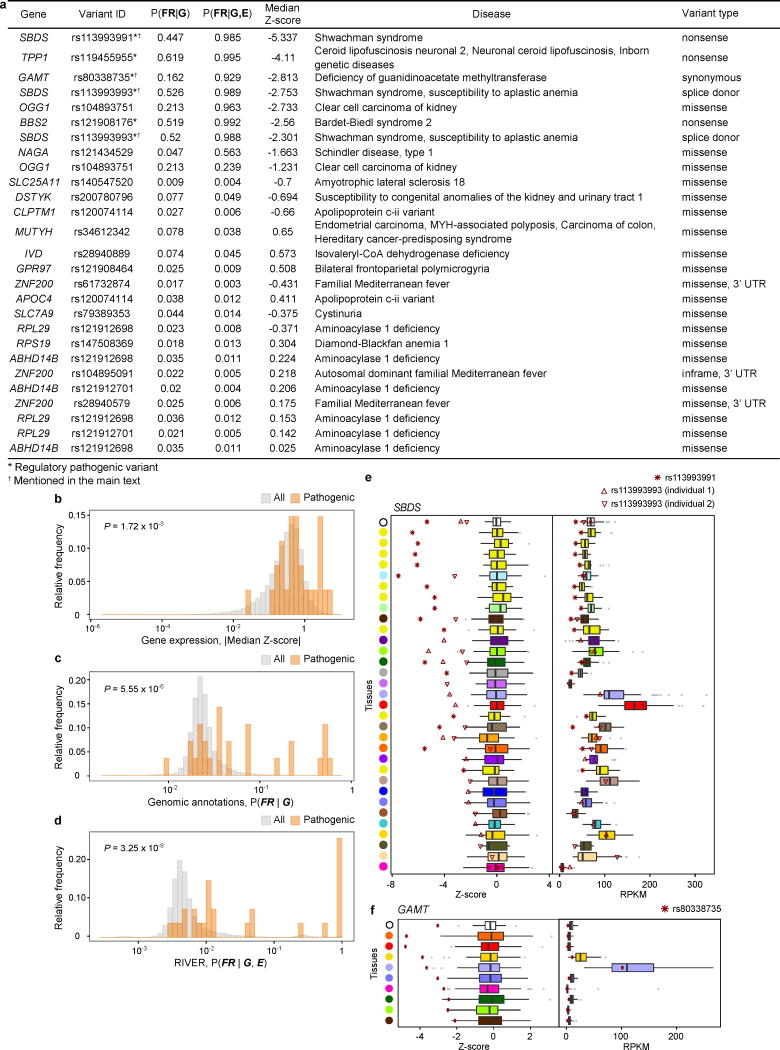
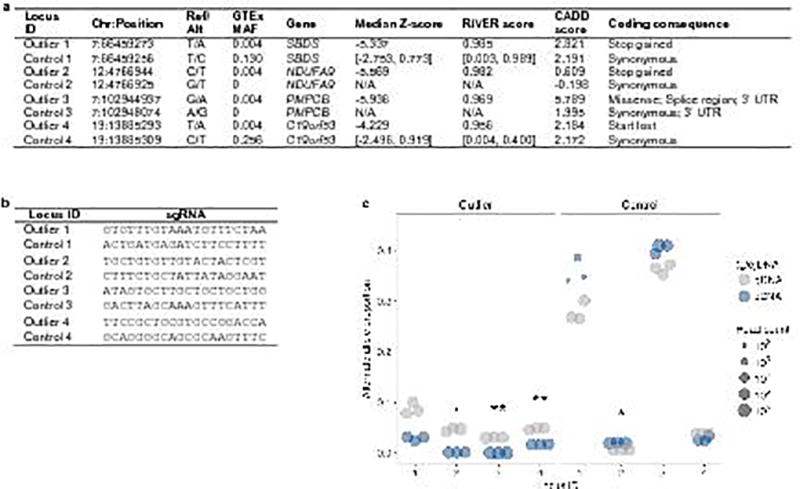
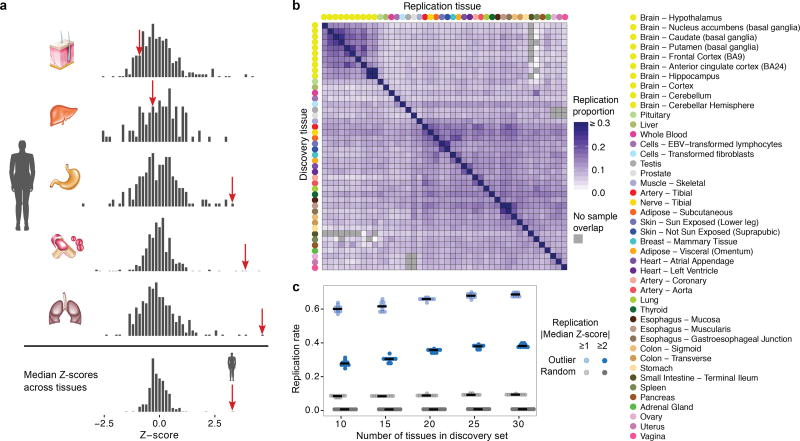
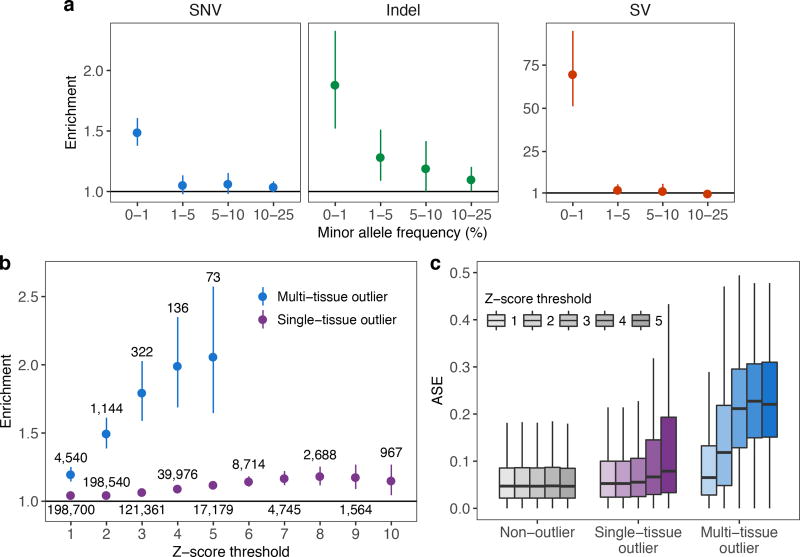
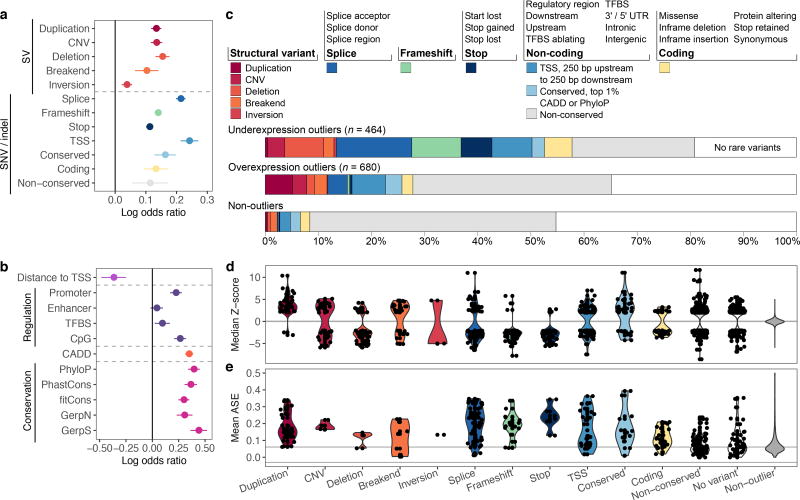
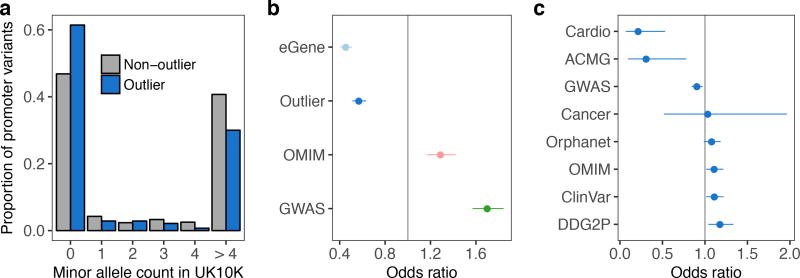
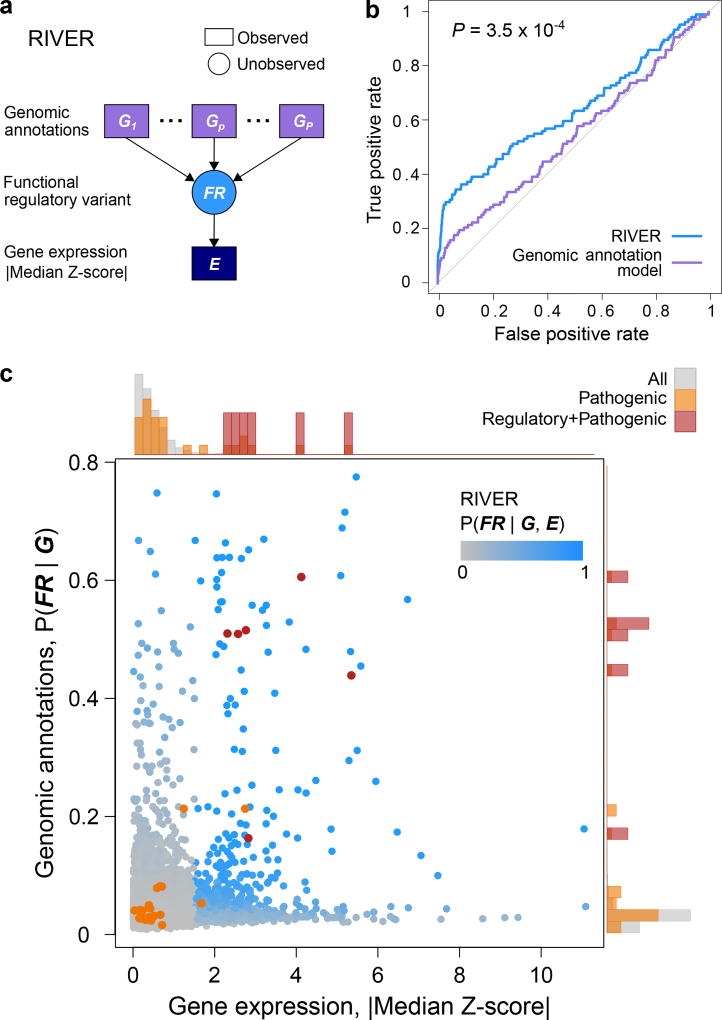
Rare genetic variants are abundant in humans and are expected to contribute to individual disease risk. While genetic association studies have successfully identified common genetic variants associated with susceptibility, these studies are not practical for identifying rare variants. Efforts to distinguish pathogenic variants from benign rare variants have leveraged the genetic code to identify deleterious protein-coding alleles, but no analogous code exists for non-coding variants. Therefore, ascertaining which rare variants have phenotypic effects remains a major challenge. Rare non-coding variants have been associated with extreme gene expression in studies using single tissues, but their effects across tissues are unknown. Here we identify gene expression outliers, or individuals showing extreme expression levels for a particular gene, across 44 human tissues by using combined analyses of whole genomes and multi-tissue RNA-sequencing data from the Genotype-Tissue Expression (GTEx) project v6p release. We find that 58% of underexpression and 28% of overexpression outliers have nearby conserved rare variants compared to 8% of non-outliers. Additionally, we developed RIVER (RNA-informed variant effect on regulation), a Bayesian statistical model that incorporates expression data to predict a regulatory effect for rare variants with higher accuracy than models using genomic annotations alone. Overall, we demonstrate that rare variants contribute to large gene expression changes across tissues and provide an integrative method for interpretation of rare variants in individual genomes.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Human genomics: Cracking the regulatory code.Nature. 2017 Oct 11;550(7675):190-191. doi: 10.1038/550190a. Nature. 2017. PMID: 29022577 No abstract available.
-
A more personal view of human-gene regulation.Nature. 2017 Oct 11;550(7675):157. doi: 10.1038/550157a. Nature. 2017. PMID: 29022932 No abstract available.
-
Gene-expression study raises thorny ethical issues.Nature. 2017 Oct 11;550(7675):169-170. doi: 10.1038/550169a. Nature. 2017. PMID: 29022940 No abstract available.
-
Gene expression: Principles of gene regulation across tissues.Nat Rev Genet. 2017 Dec;18(12):701. doi: 10.1038/nrg.2017.94. Epub 2017 Nov 7. Nat Rev Genet. 2017. PMID: 29109523 No abstract available.
-
Taking genomics research to the next level: The Genotype-Tissue expression project.Mov Disord. 2018 Jul;33(7):1097. doi: 10.1002/mds.27445. Mov Disord. 2018. PMID: 30153387 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
