

Comparison of classical multi-locus sequence typing software for next-generation sequencing data
- PMID: 29026660
- PMCID: PMC5610716
- DOI: 10.1099/mgen.0.000124
Comparison of classical multi-locus sequence typing software for next-generation sequencing data
Abstract
Multi-locus sequence typing (MLST) is a widely used method for categorizing bacteria. Increasingly, MLST is being performed using next-generation sequencing (NGS) data by reference laboratories and for clinical diagnostics. Many software applications have been developed to calculate sequence types from NGS data; however, there has been no comprehensive review to date on these methods. We have compared eight of these applications against real and simulated data, and present results on: (1) the accuracy of each method against traditional typing methods, (2) the performance on real outbreak datasets, (3) the impact of contamination and varying depth of coverage, and (4) the computational resource requirements.
Keywords: MLST; multi-locus sequence typing; next-generation sequencing; software comparison.
Figures
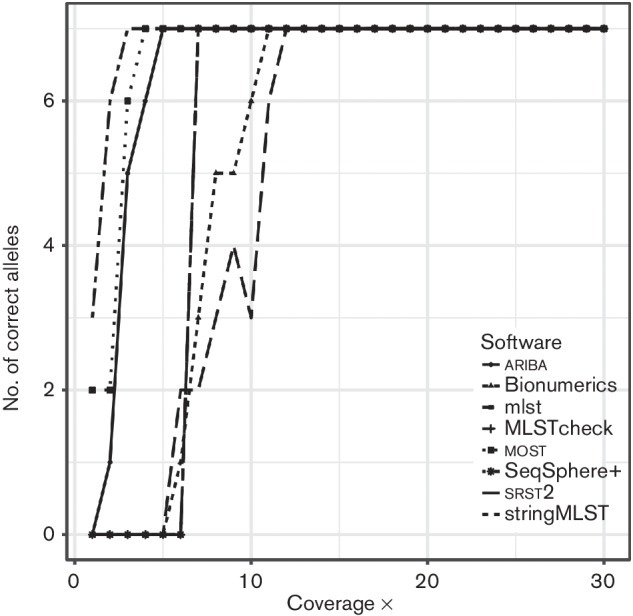
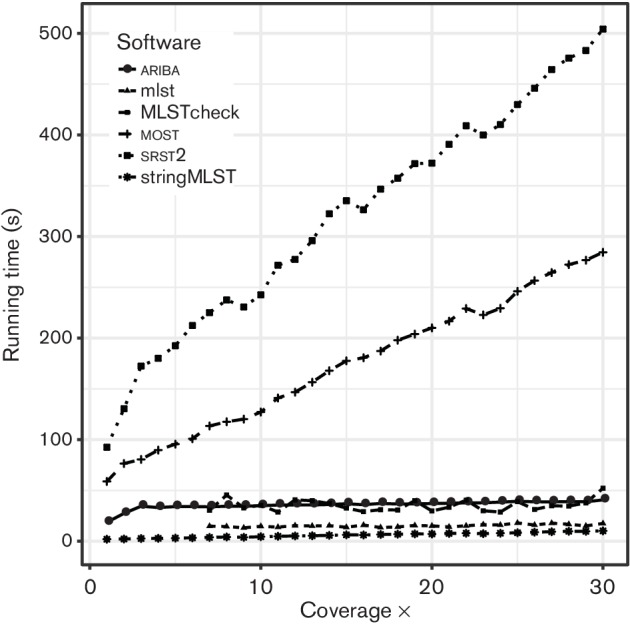
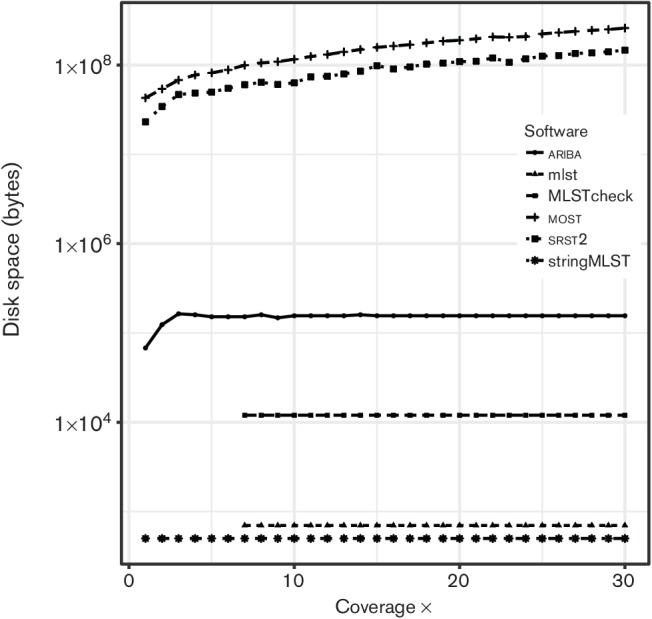
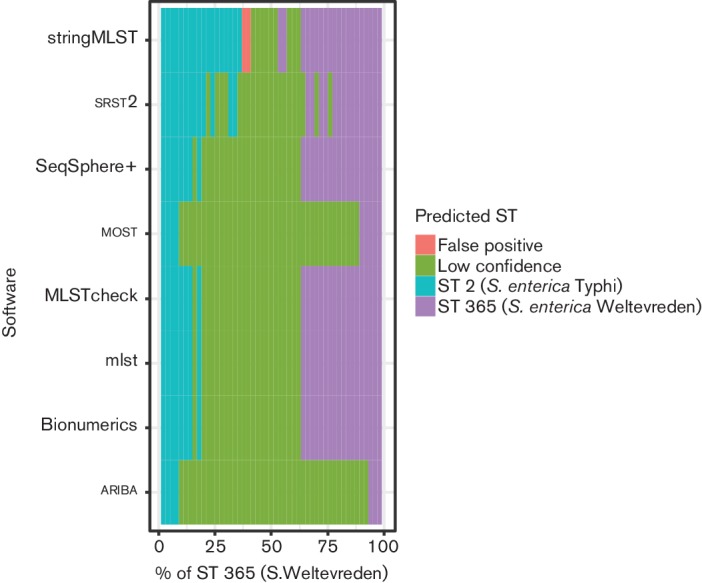
References
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources