

ER Protein Quality Control and the Unfolded Protein Response in the Heart
- PMID: 29026925
- PMCID: PMC6459409
- DOI: 10.1007/82_2017_54
ER Protein Quality Control and the Unfolded Protein Response in the Heart
Abstract
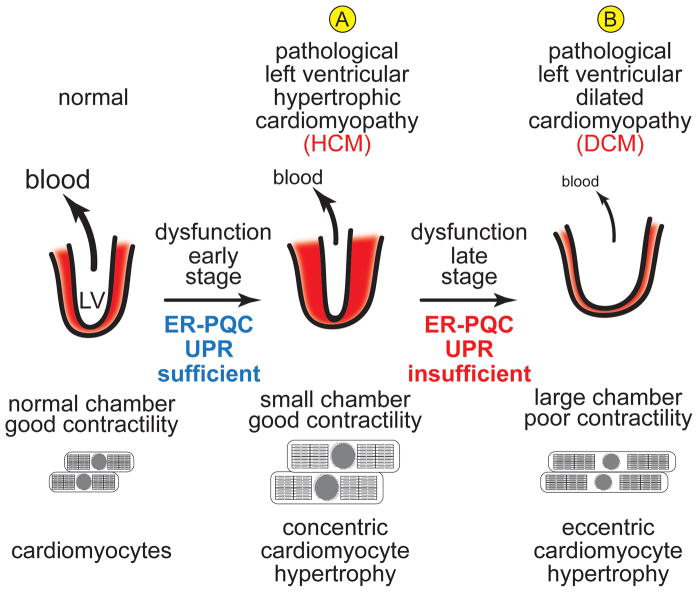
Cardiac myocytes are the cells responsible for the robust ability of the heart to pump blood throughout the circulatory system. Cardiac myocytes grow in response to a variety of physiological and pathological conditions; this growth challenges endoplasmic reticulum-protein quality control (ER-PQC), a major feature of which includes the unfolded protein response (UPR). ER-PQC and the UPR in cardiac myocytes growing under physiological conditions, including normal development, exercise, and pregnancy, are sufficient to support hypertrophic growth of each cardiac myocyte. However, the ER-PQC and UPR are insufficient to respond to the challenge of cardiac myocyte growth under pathological conditions, including myocardial infarction and heart failure. In part, this insufficiency is due to a continual decline in the expression levels of important adaptive UPR components as a function of age and during myocardial pathology. This chapter will discuss the physiological and pathological conditions unique to the heart that involves ER-PQC, and whether the UPR is adaptive or maladaptive under these circumstances.
Figures





References
-
- Belmont PJ, Tadimalla A, Chen WJ, Martindale JJ, Thuerauf DJ, Marcinko M, Gude N, Sussman MA, Glembotski CC. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J Biol Chem. 2008;283(20):14012–14021. doi: 10.1074/jbc.M709776200. M709776200 [pii] - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
