

Comparative analysis of differential gene expression tools for RNA sequencing time course data
- PMID: 29028903
- PMCID: PMC6357553
- DOI: 10.1093/bib/bbx115
Comparative analysis of differential gene expression tools for RNA sequencing time course data
Abstract
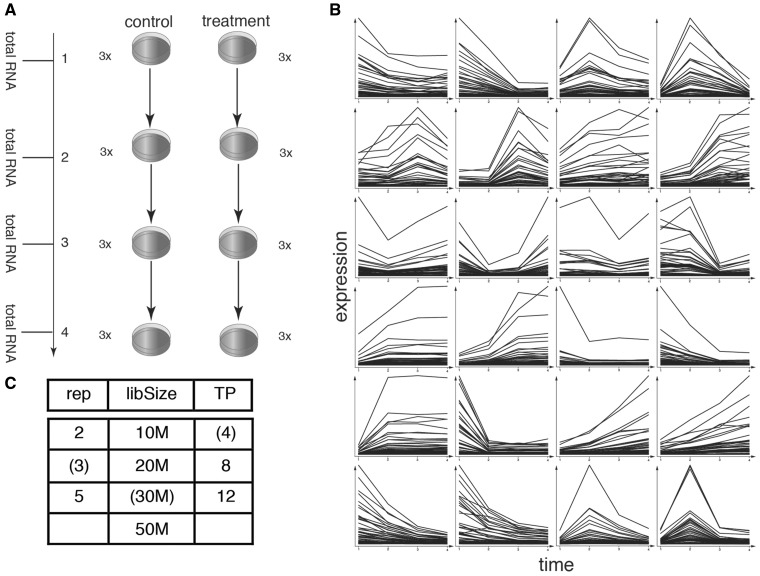
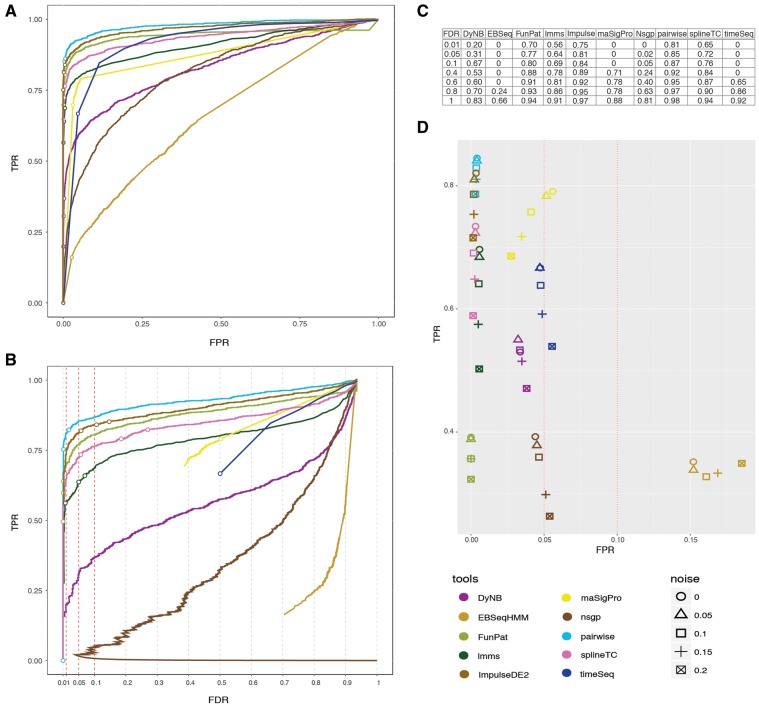
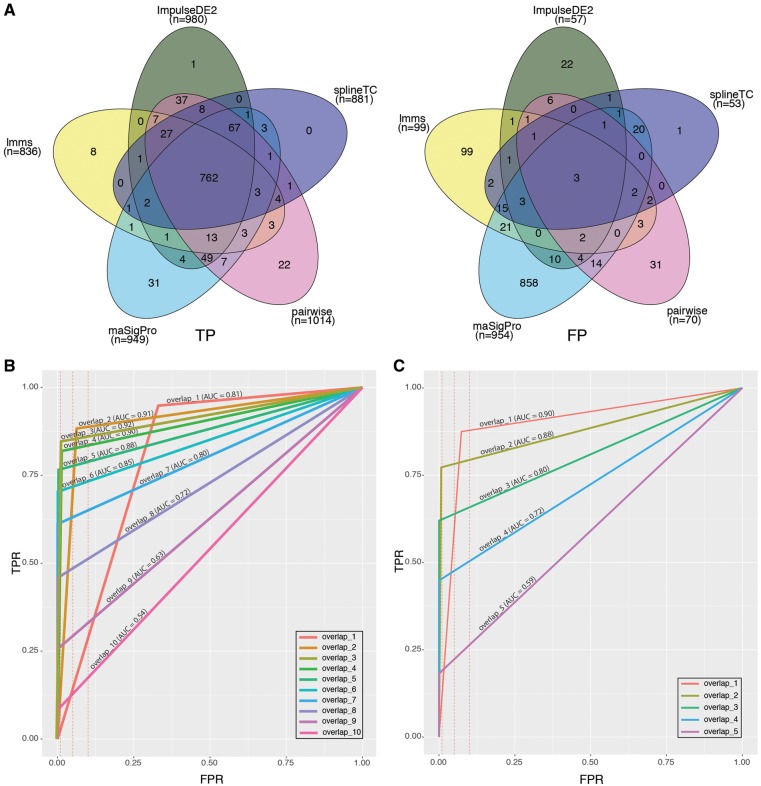
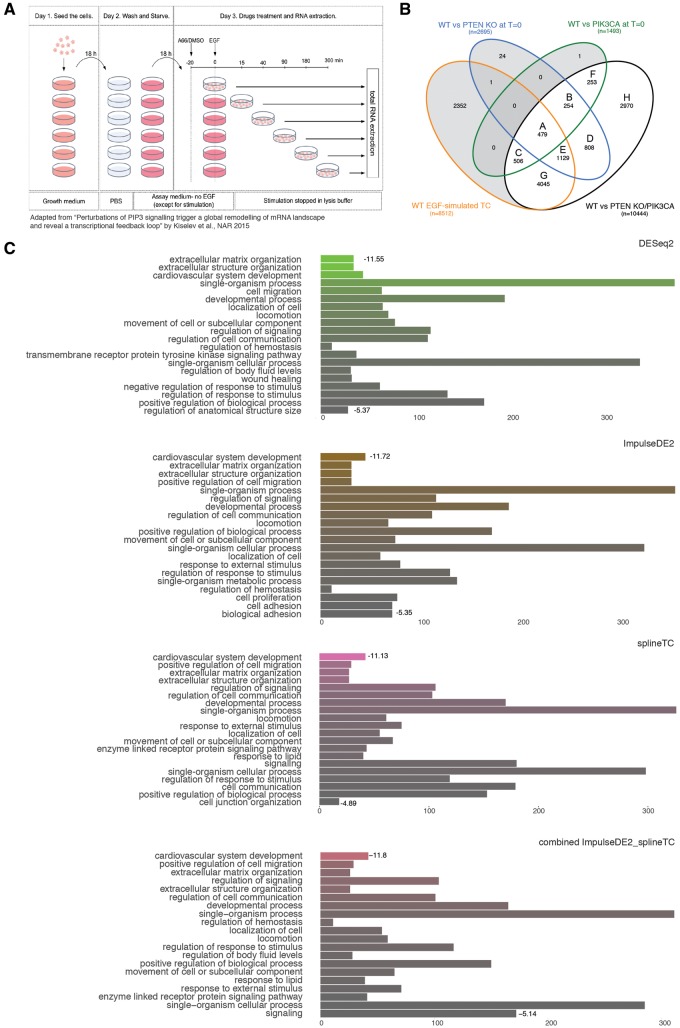
RNA sequencing (RNA-seq) has become a standard procedure to investigate transcriptional changes between conditions and is routinely used in research and clinics. While standard differential expression (DE) analysis between two conditions has been extensively studied, and improved over the past decades, RNA-seq time course (TC) DE analysis algorithms are still in their early stages. In this study, we compare, for the first time, existing TC RNA-seq tools on an extensive simulation data set and validated the best performing tools on published data. Surprisingly, TC tools were outperformed by the classical pairwise comparison approach on short time series (<8 time points) in terms of overall performance and robustness to noise, mostly because of high number of false positives, with the exception of ImpulseDE2. Overlapping of candidate lists between tools improved this shortcoming, as the majority of false-positive, but not true-positive, candidates were unique for each method. On longer time series, pairwise approach was less efficient on the overall performance compared with splineTC and maSigPro, which did not identify any false-positive candidate.
Figures
References
-
- do Amaral MN, Arge LW, Benitez LC. Comparative transcriptomics of rice plants under cold, iron, and salt stresses. Funct Integr Genomics 2016;16:567–79. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
