

A retractable lid in lecithin:cholesterol acyltransferase provides a structural mechanism for activation by apolipoprotein A-I
- PMID: 29030428
- PMCID: PMC5724016
- DOI: 10.1074/jbc.M117.802736
A retractable lid in lecithin:cholesterol acyltransferase provides a structural mechanism for activation by apolipoprotein A-I
Abstract
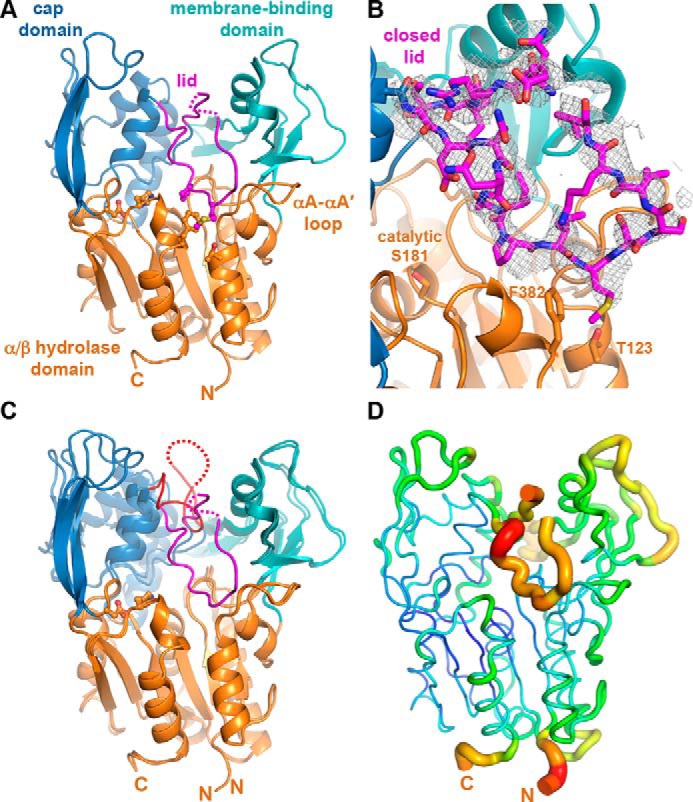
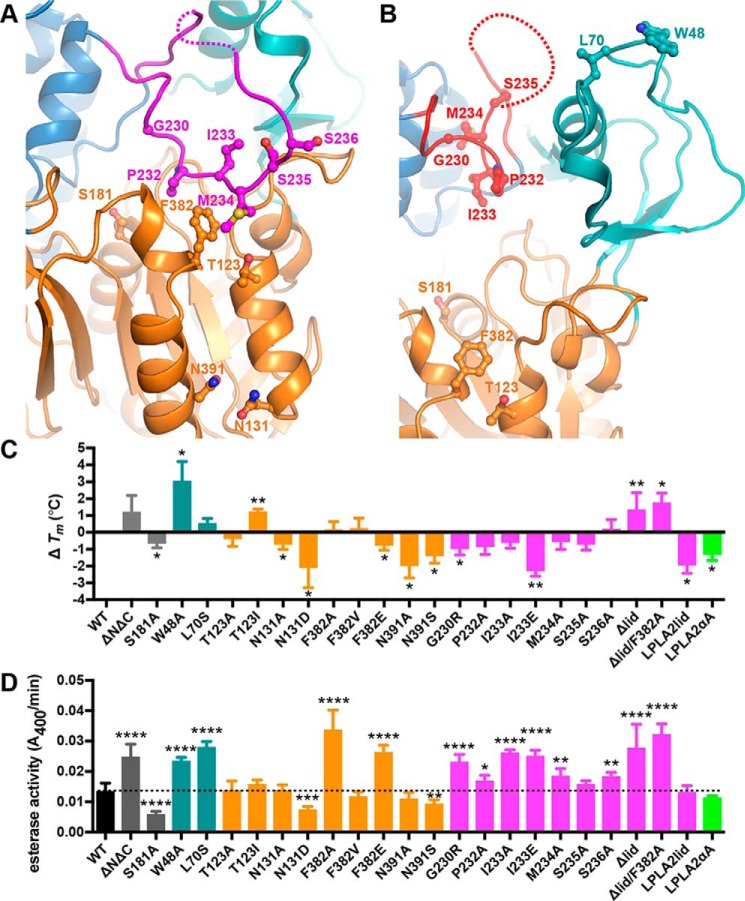
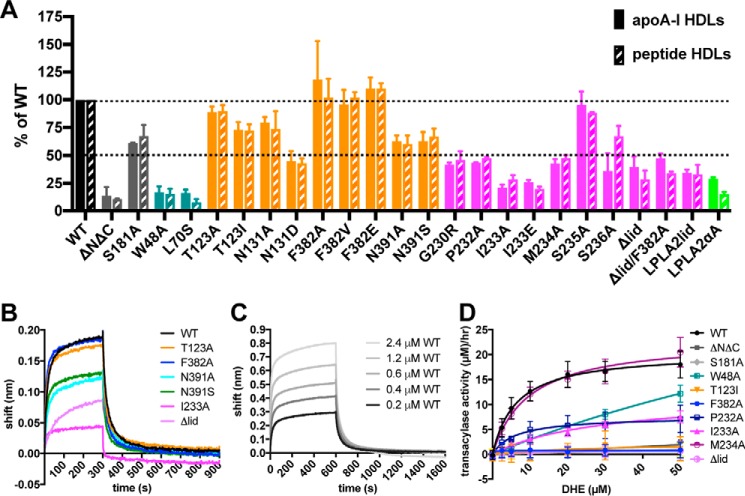
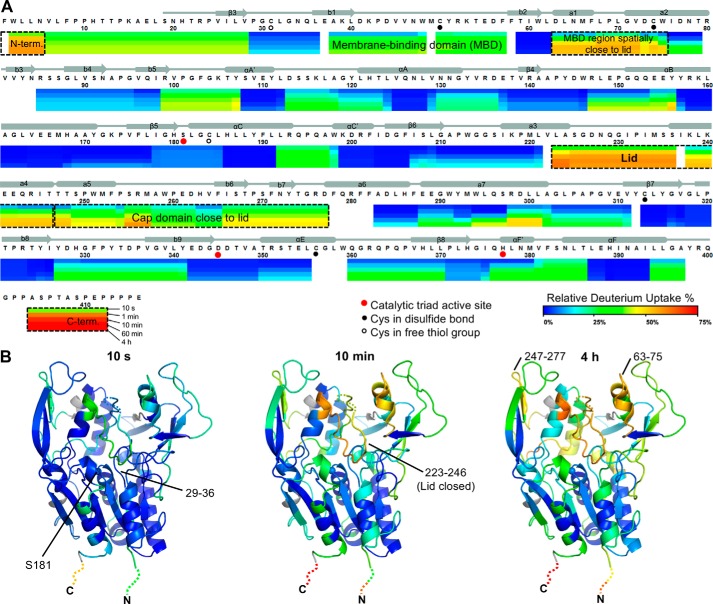
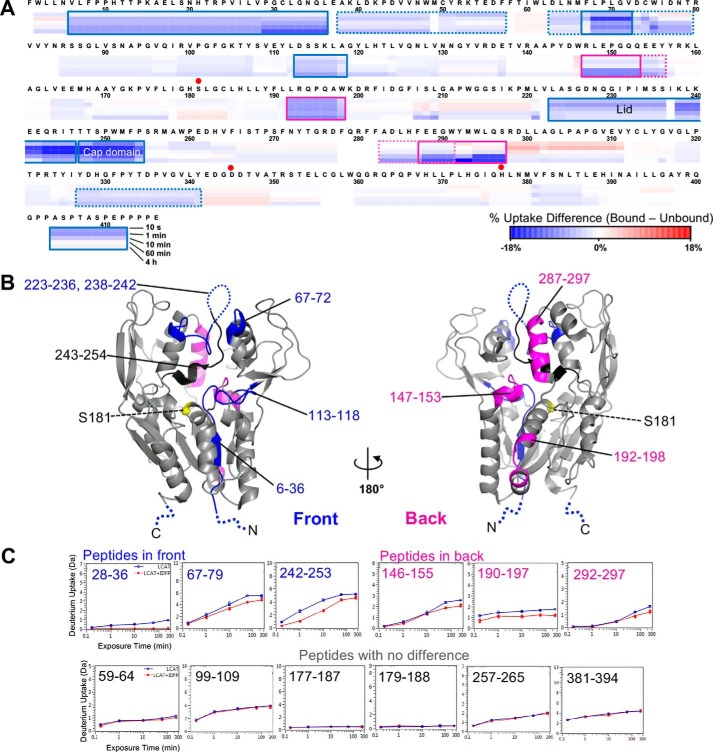
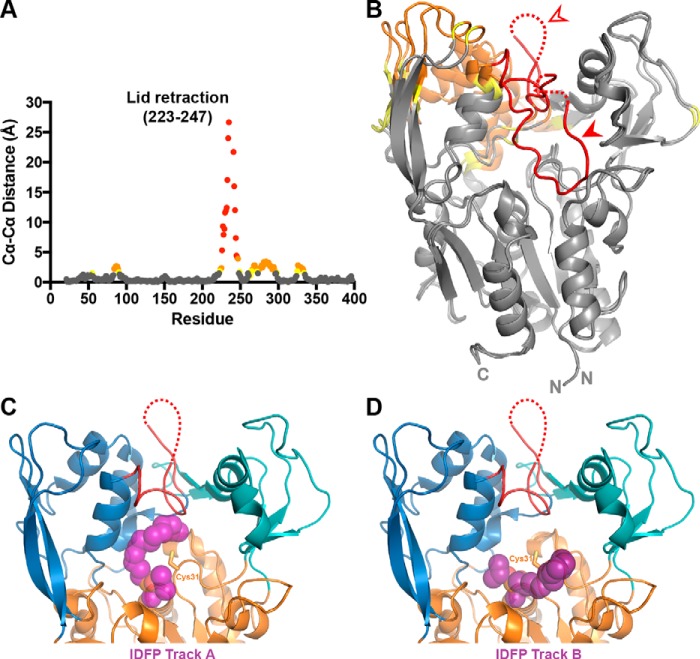
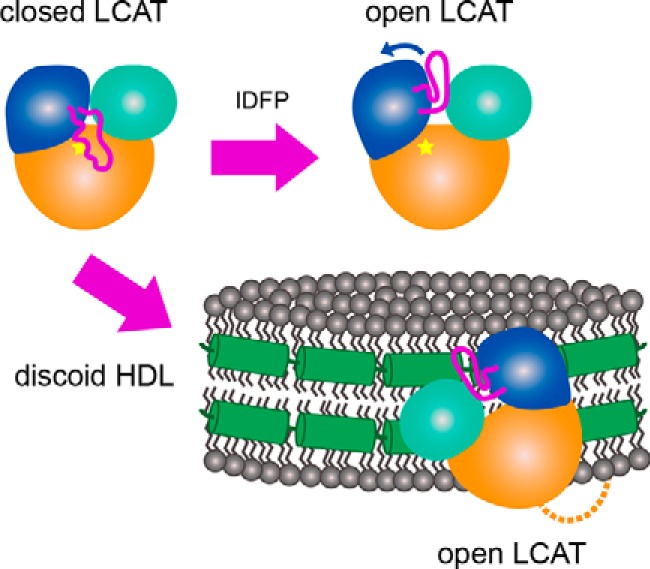
Lecithin:cholesterol acyltransferase (LCAT) plays a key role in reverse cholesterol transport by transferring an acyl group from phosphatidylcholine to cholesterol, promoting the maturation of high-density lipoproteins (HDL) from discoidal to spherical particles. LCAT is activated through an unknown mechanism by apolipoprotein A-I (apoA-I) and other mimetic peptides that form a belt around HDL. Here, we report the crystal structure of LCAT with an extended lid that blocks access to the active site, consistent with an inactive conformation. Residues Thr-123 and Phe-382 in the catalytic domain form a latch-like interaction with hydrophobic residues in the lid. Because these residues are mutated in genetic disease, lid displacement was hypothesized to be an important feature of apoA-I activation. Functional studies of site-directed mutants revealed that loss of latch interactions or the entire lid enhanced activity against soluble ester substrates, and hydrogen-deuterium exchange (HDX) mass spectrometry revealed that the LCAT lid is extremely dynamic in solution. Upon addition of a covalent inhibitor that mimics one of the reaction intermediates, there is an overall decrease in HDX in the lid and adjacent regions of the protein, consistent with ordering. These data suggest a model wherein the active site of LCAT is shielded from soluble substrates by a dynamic lid until it interacts with HDL to allow transesterification to proceed.
Keywords: HDX MS; acyltransferase; apolipoprotein; cholesterol; conformational change; crystallography; high-density lipoprotein (HDL); inhibitor; lecithin; structural biology.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
J. A., C. L., and M. J. A. declare that they are employees of MedImmune, the global biologics R&D arm of AstraZeneca, and own stock/stock options in AstraZeneca
Figures
References
-
- Calabresi L., Simonelli S., Gomaraschi M., and Franceschini G. (2012) Genetic lecithin:cholesterol acyltransferase deficiency and cardiovascular disease. Atherosclerosis 222, 299–306 - PubMed
-
- Glomset J. A. (1968) The plasma lecithins:cholesterol acyltransferase reaction. J. Lipid Res. 9, 155–167 - PubMed
-
- Chen C. H., and Albers J. J. (1982) Distribution of lecithin-cholesterol acyltransferase (LCAT) in human plasma lipoprotein fractions. Evidence for the association of active LCAT with low-density lipoproteins. Biochem. Biophys. Res. Commun. 107, 1091–1096 - PubMed
-
- Zhao Y., Thorngate F. E., Weisgraber K. H., Williams D. L., and Parks J. S. (2005) Apolipoprotein E is the major physiological activator of lecithin-cholesterol acyltransferase (LCAT) on apolipoprotein B lipoproteins. Biochemistry 44, 1013–1025 - PubMed
-
- Kuivenhoven J. A., Pritchard H., Hill J., Frohlich J., Assmann G., and Kastelein J. (1997) The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J. Lipid Res. 38, 191–205 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
