

Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer's disease
- PMID: 29030541
- PMCID: PMC5714957
- DOI: 10.1038/s41467-017-00867-z
Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer's disease
Erratum in
-
Publisher Correction: Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer's disease.Nat Commun. 2018 Oct 31;9(1):4625. doi: 10.1038/s41467-018-07153-6. Nat Commun. 2018. PMID: 30382106 Free PMC article.
Abstract
The abundance of mRNA is mainly determined by the rates of RNA transcription and decay. Here, we present a method for unbiased estimation of differential mRNA decay rate from RNA-sequencing data by modeling the kinetics of mRNA metabolism. We show that in all primary human tissues tested, and particularly in the central nervous system, many pathways are regulated at the mRNA stability level. We present a parsimonious regulatory model consisting of two RNA-binding proteins and four microRNAs that modulate the mRNA stability landscape of the brain, which suggests a new link between RBFOX proteins and Alzheimer's disease. We show that downregulation of RBFOX1 leads to destabilization of mRNAs encoding for synaptic transmission proteins, which may contribute to the loss of synaptic function in Alzheimer's disease. RBFOX1 downregulation is more likely to occur in older and female individuals, consistent with the association of Alzheimer's disease with age and gender."mRNA abundance is determined by the rates of transcription and decay. Here, the authors propose a method for estimating the rate of differential mRNA decay from RNA-seq data and model mRNA stability in the brain, suggesting a link between mRNA stability and Alzheimer's disease."
Conflict of interest statement
The authors declare no competing financial interests.
Figures
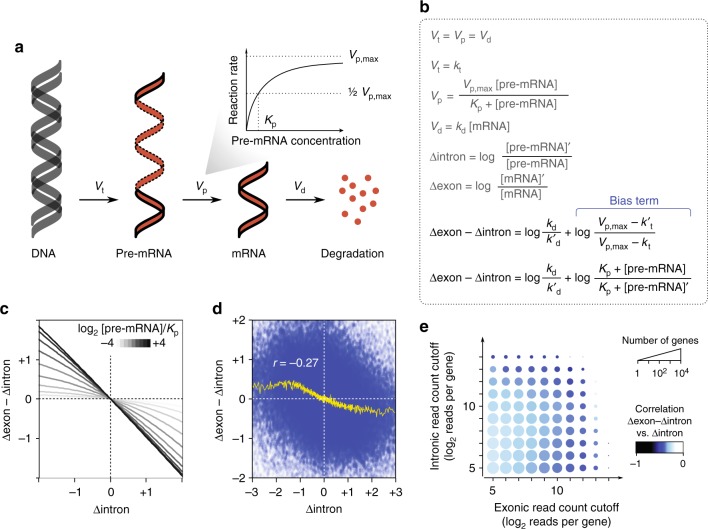
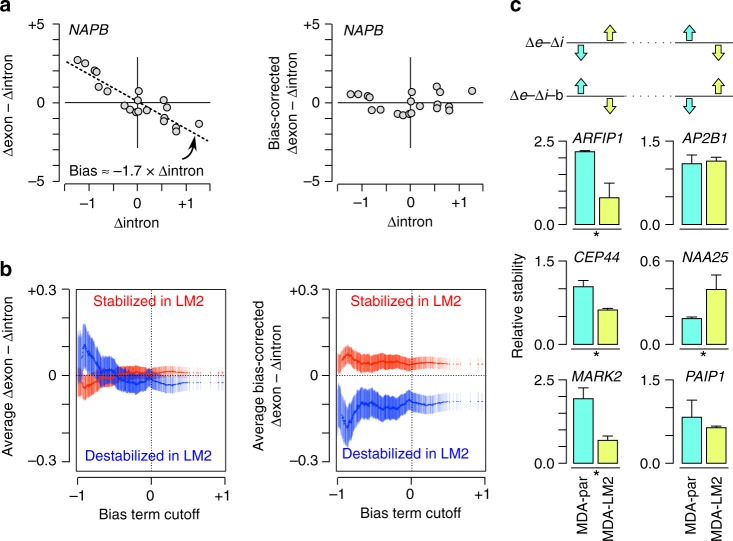
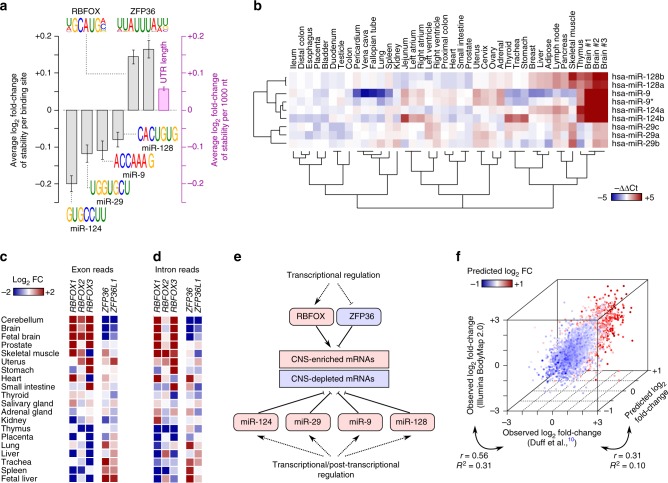


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
