

Signatures of positive selection reveal a universal role of chromatin modifiers as cancer driver genes
- PMID: 29030609
- PMCID: PMC5640613
- DOI: 10.1038/s41598-017-12888-1
Signatures of positive selection reveal a universal role of chromatin modifiers as cancer driver genes
Abstract
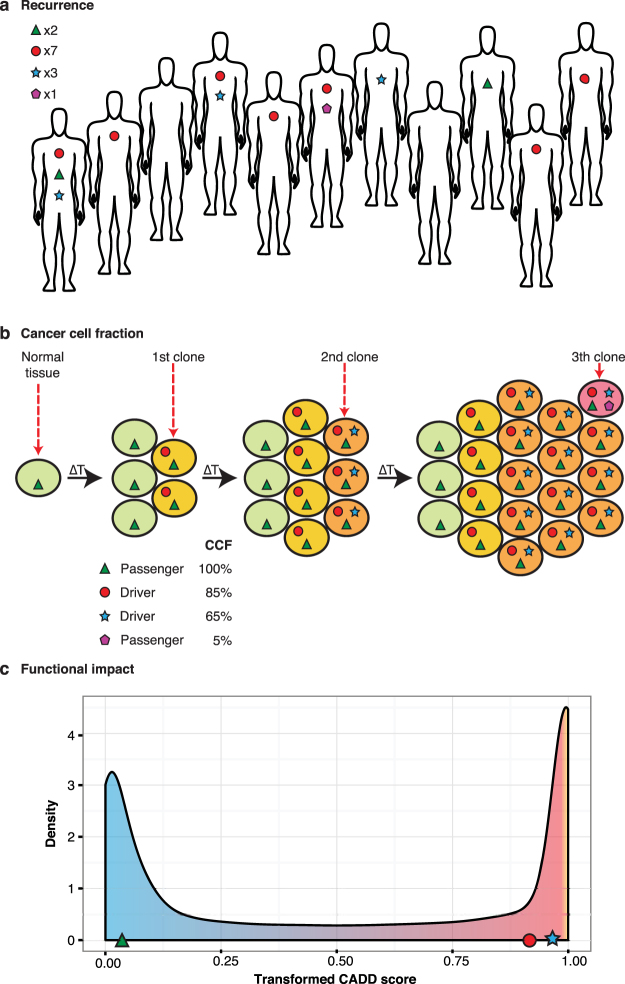
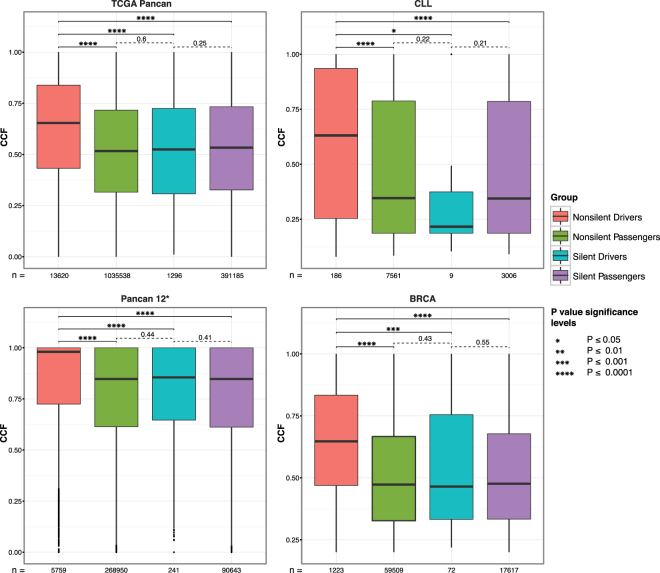
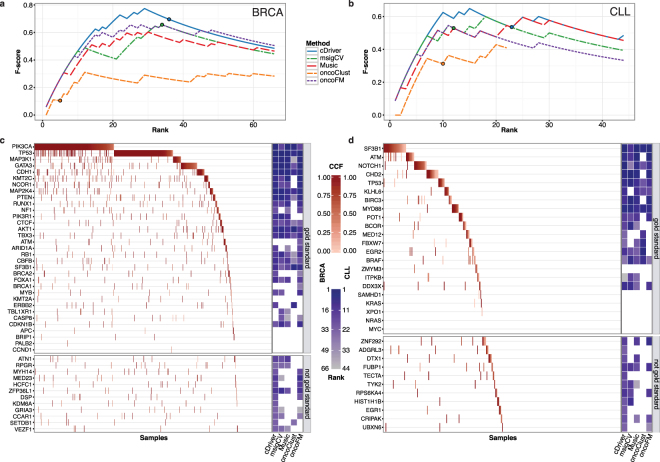
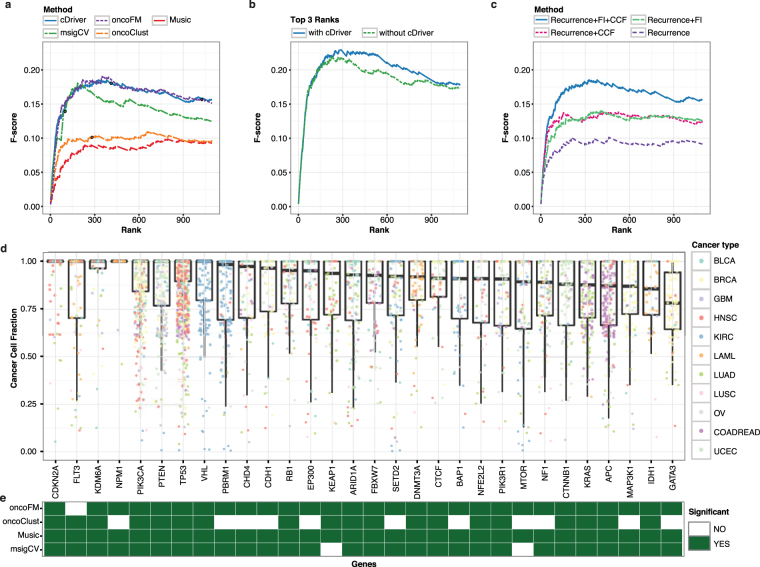
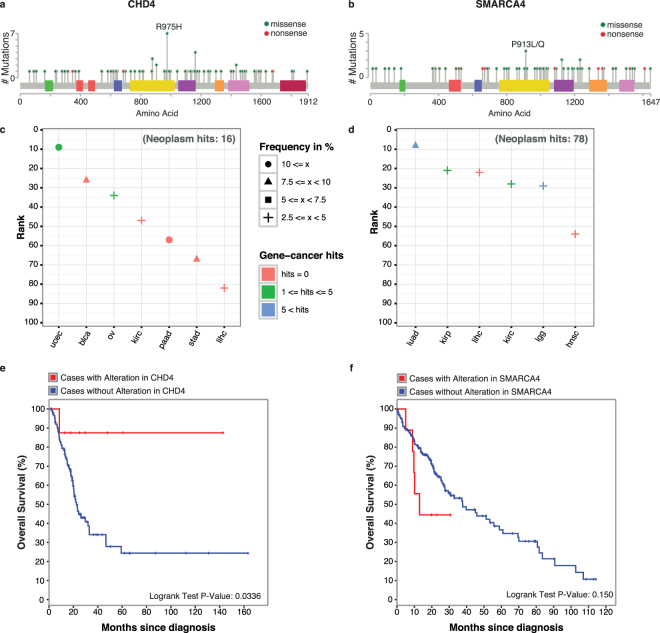
Tumors are composed of an evolving population of cells subjected to tissue-specific selection, which fuels tumor heterogeneity and ultimately complicates cancer driver gene identification. Here, we integrate cancer cell fraction, population recurrence, and functional impact of somatic mutations as signatures of selection into a Bayesian model for driver prediction. We demonstrate that our model, cDriver, outperforms competing methods when analyzing solid tumors, hematological malignancies, and pan-cancer datasets. Applying cDriver to exome sequencing data of 21 cancer types from 6,870 individuals revealed 98 unreported tumor type-driver gene connections. These novel connections are highly enriched for chromatin-modifying proteins, hinting at a universal role of chromatin regulation in cancer etiology. Although infrequently mutated as single genes, we show that chromatin modifiers are altered in a large fraction of cancer patients. In summary, we demonstrate that integration of evolutionary signatures is key for identifying mutational driver genes, thereby facilitating the discovery of novel therapeutic targets for cancer treatment.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
