

Inhibition of cholinergic potentiation of insulin secretion from pancreatic islets by chronic elevation of glucose and fatty acids: Protection by casein kinase 2 inhibitor
- PMID: 29031723
- PMCID: PMC5641685
- DOI: 10.1016/j.molmet.2017.07.017
Inhibition of cholinergic potentiation of insulin secretion from pancreatic islets by chronic elevation of glucose and fatty acids: Protection by casein kinase 2 inhibitor
Abstract
Objectives: Chronic hyperlipidemia and hyperglycemia are characteristic features of type 2 diabetes (T2DM) that are thought to cause or contribute to β-cell dysfunction by "glucolipotoxicity." Previously we have shown that acute treatment of pancreatic islets with fatty acids (FA) decreases acetylcholine-potentiated insulin secretion. This acetylcholine response is mediated by M3 muscarinic receptors, which play a key role in regulating β-cell function. Here we examine whether chronic FA exposure also inhibits acetylcholine-potentiated insulin secretion using mouse and human islets.
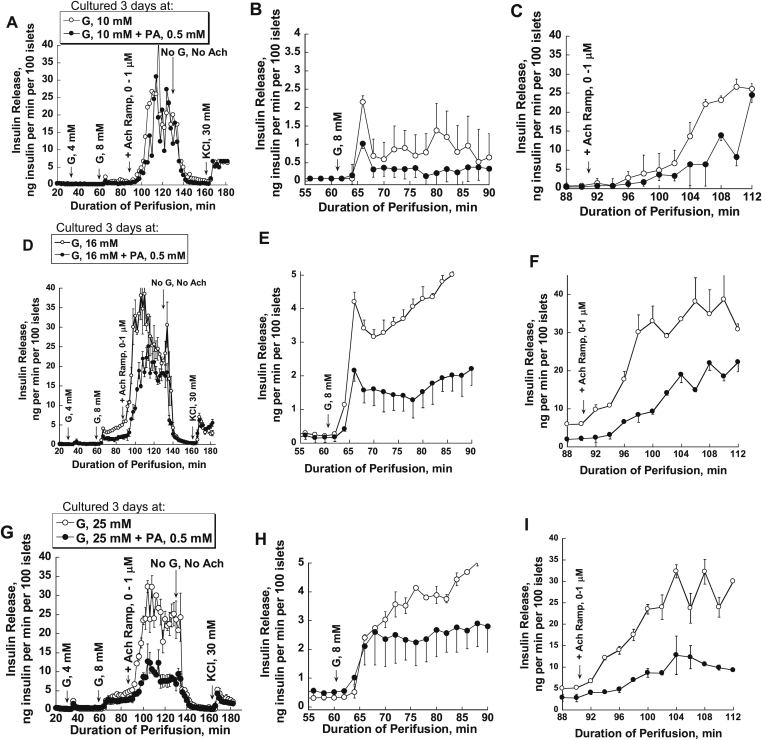
Methods: Islets were cultured for 3 or 4 days at different glucose concentration with 0.5 mM palmitic acid (PA) or a 2:1 mixture of PA and oleic acid (OA) at 1% albumin (PA/BSA molar ratio 3.3). Afterwards, the response to glucose and acetylcholine were studied in perifusion experiments.
Results: FA-induced impairment of insulin secretion and Ca2+ signaling depended strongly on the glucose concentrations of the culture medium. PA and OA in combination reduced acetylcholine potentiation of insulin secretion more than PA alone, both in mouse and human islets, with no evidence of a protective role of OA. In contrast, lipotoxicity was not observed with islets cultured for 3 days in medium containing less than 1 mM glucose and a mixture of glutamine and leucine (7 mM each). High glucose and FAs reduced endoplasmic reticulum (ER) Ca2+ storage capacity; however, preserving ER Ca2+ by blocking the IP3 receptor with xestospongin C did not protect islets from glucolipotoxic effects on insulin secretion. In contrast, an inhibitor of casein kinase 2 (CK2) protected the glucose dependent acetylcholine potentiation of insulin secretion in mouse and human islets against glucolipotoxicity.
Conclusions: These results show that chronic FA treatment decreases acetylcholine potentiation of insulin secretion and that this effect is strictly glucose dependent and might involve CK2 phosphorylation of β-cell M3 muscarinic receptors.
Keywords: Acetylcholine; Fatty acids; Glucolipotoxicity; Insulin secretion; Pancreatic islets.
Copyright © 2017 The Authors. Published by Elsevier GmbH.. All rights reserved.
Figures







Similar articles
-
Palmitic acid acutely inhibits acetylcholine- but not GLP-1-stimulated insulin secretion in mouse pancreatic islets.Am J Physiol Endocrinol Metab. 2010 Sep;299(3):E475-85. doi: 10.1152/ajpendo.00072.2010. Epub 2010 Jul 6. Am J Physiol Endocrinol Metab. 2010. PMID: 20606076 Free PMC article.
-
Oleic acid protects insulin-secreting INS-1E cells against palmitic acid-induced lipotoxicity along with an amelioration of ER stress.Endocrine. 2019 Jun;64(3):512-524. doi: 10.1007/s12020-019-01867-3. Epub 2019 Feb 18. Endocrine. 2019. PMID: 30778898
-
Mechanisms of octanoic acid potentiation of insulin secretion in isolated islets.Islets. 2019;11(4):77-88. doi: 10.1080/19382014.2019.1566683. Epub 2019 Mar 8. Islets. 2019. PMID: 30849280 Free PMC article.
-
Regulatory Role of Fatty Acid Metabolism on Glucose-Induced Changes in Insulin and Glucagon Secretion by Pancreatic Islet Cells.Int J Mol Sci. 2024 May 31;25(11):6052. doi: 10.3390/ijms25116052. Int J Mol Sci. 2024. PMID: 38892240 Free PMC article. Review.
-
Signal transduction in pancreatic beta-cells: regulation of insulin secretion by information flow in the phospholipase C/protein kinase C pathway.Front Biosci. 1997 Mar 15;2:d160-72. doi: 10.2741/a180. Front Biosci. 1997. PMID: 9159224 Review.
Cited by
-
Protein kinase CK2: a potential therapeutic target for diverse human diseases.Signal Transduct Target Ther. 2021 May 17;6(1):183. doi: 10.1038/s41392-021-00567-7. Signal Transduct Target Ther. 2021. PMID: 33994545 Free PMC article. Review.
-
Targeting Protein Kinases to Protect Beta-Cell Function and Survival in Diabetes.Int J Mol Sci. 2024 Jun 11;25(12):6425. doi: 10.3390/ijms25126425. Int J Mol Sci. 2024. PMID: 38928130 Free PMC article. Review.
-
Nrf2 Activation Protects Mouse Beta Cells from Glucolipotoxicity by Restoring Mitochondrial Function and Physiological Redox Balance.Oxid Med Cell Longev. 2019 Nov 11;2019:7518510. doi: 10.1155/2019/7518510. eCollection 2019. Oxid Med Cell Longev. 2019. PMID: 31827698 Free PMC article.
-
Gq signaling in α cells is critical for maintaining euglycemia.JCI Insight. 2021 Dec 22;6(24):e152852. doi: 10.1172/jci.insight.152852. JCI Insight. 2021. PMID: 34752420 Free PMC article.
-
Blocking Kir6.2 channels with SpTx1 potentiates glucose-stimulated insulin secretion from murine pancreatic β cells and lowers blood glucose in diabetic mice.Elife. 2022 Feb 25;11:e77026. doi: 10.7554/eLife.77026. Elife. 2022. PMID: 35212627 Free PMC article.
References
-
- Ahren B. Autonomic regulation of islet hormone secretion – implications for health and disease. Diabetologia. 2000;43:393–410. - PubMed
-
- Ahren B., Sauerberg P., Thomsen C. Increased insulin secretion and normalization of glucose tolerance by cholinergic agonism in high fat-fed mice. American Journal of Physiology. 1999;277:E93–E102. - PubMed
-
- Branstrom R., Aspinwall C.A., Valimaki S., Ostensson C.G., Tibell A., Eckhard M. Long-chain CoA esters activate human pancreatic beta-cell KATP channels: potential role in Type 2 diabetes. Diabetologia. 2004;47:277–283. - PubMed
-
- Branstrom R., Corkey B.E., Berggren P.O., Larsson O. Evidence for a unique long chain acyl-CoA ester binding site on the ATP-regulated potassium channel in mouse pancreatic beta cells. The Journal of Biological Chemistry. 1997;272:17390–17394. - PubMed
-
- Carpentier A., Giacca A., Lewis G.F. Effect of increased plasma non-esterified fatty acids (NEFAs) on arginine-stimulated insulin secretion in obese humans. Diabetologia. 2001;44:1989–1997. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
