

Genetics of Pulmonary Arterial Hypertension
- PMID: 29032562
- PMCID: PMC7722263
- DOI: 10.1055/s-0037-1606201
Genetics of Pulmonary Arterial Hypertension
Abstract
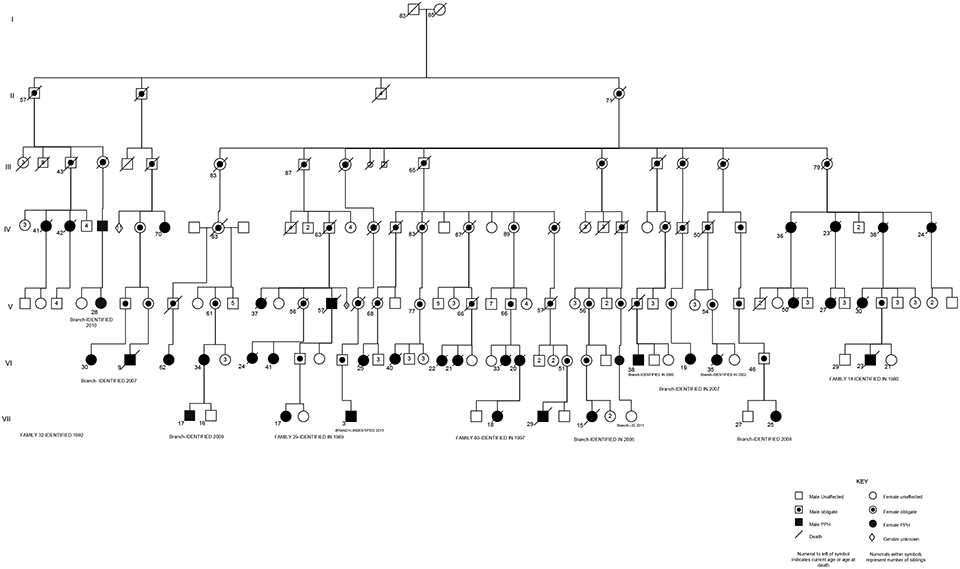
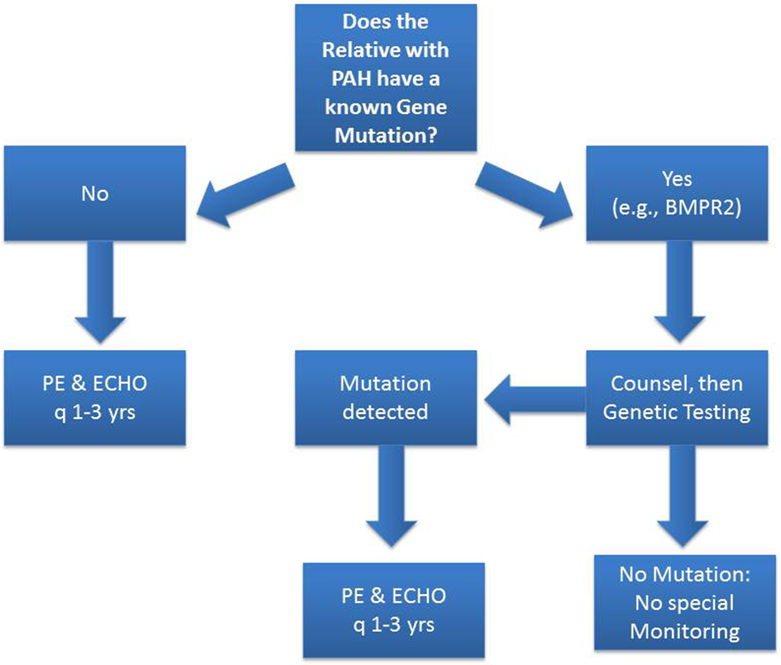
Tremendous progress has been made in understanding the genetics of pulmonary arterial hypertension (PAH) since its description in the 1950s as a primary disorder of the pulmonary vasculature. Heterozygous germline mutations in the gene coding bone morphogenetic receptor type 2 (BMPR2) are detectable in the majority of cases of heritable PAH, and in approximately 20% of cases of idiopathic pulmonary arterial hypertension (IPAH). However, recent advances in gene discovery methods have facilitated the discovery of additional genes with mutations among those with and without familial PAH. Heritable PAH is an autosomal dominant disease characterized by reduced penetrance, variable expressivity, and female predominance. Biallelic germline mutations in the gene EIF2AK4 are now associated with pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. Growing genetic knowledge enhances our capacity to pursue and provide genetic counseling, although the issue remains complex given that the majority of carriers of PAH-related mutations will never be diagnosed with the disease.
Conflict of interest statement
Disclosure The authors report no conflicts of interest in this work.
Figures
References
-
- Dresdale DT, Schultz M, Michtom RJ. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am J Med 1951;11(06):686–705 - PubMed
-
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62(25, Suppl):D34–D41 - PubMed
-
- Loyd JE, Primm RK, Newman JH. Familial primary pulmonary hypertension: clinical patterns. Am Rev Respir Dis 1984;129(01):194–197 - PubMed
-
- Soubrier F, Chung WK, Machado R, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 2013;62(25, Suppl):D13–D21 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
