

Atypical PKC, PKCλ/ι, activates β-secretase and increases Aβ1-40/42 and phospho-tau in mouse brain and isolated neuronal cells, and may link hyperinsulinemia and other aPKC activators to development of pathological and memory abnormalities in Alzheimer's disease
- PMID: 29032894
- PMCID: PMC5705272
- DOI: 10.1016/j.neurobiolaging.2017.09.001
Atypical PKC, PKCλ/ι, activates β-secretase and increases Aβ1-40/42 and phospho-tau in mouse brain and isolated neuronal cells, and may link hyperinsulinemia and other aPKC activators to development of pathological and memory abnormalities in Alzheimer's disease
Abstract
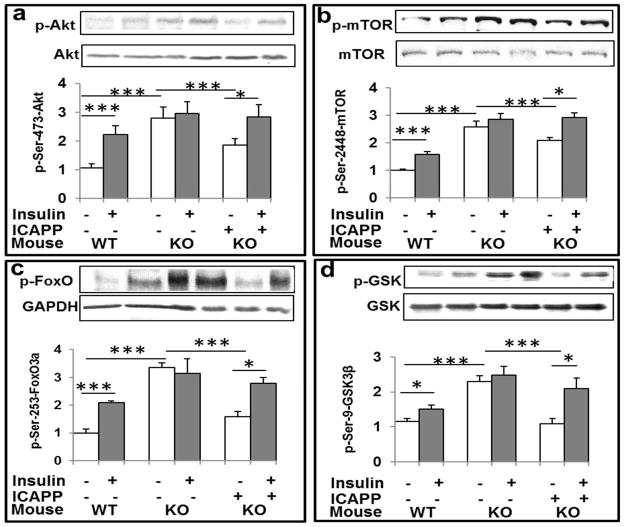
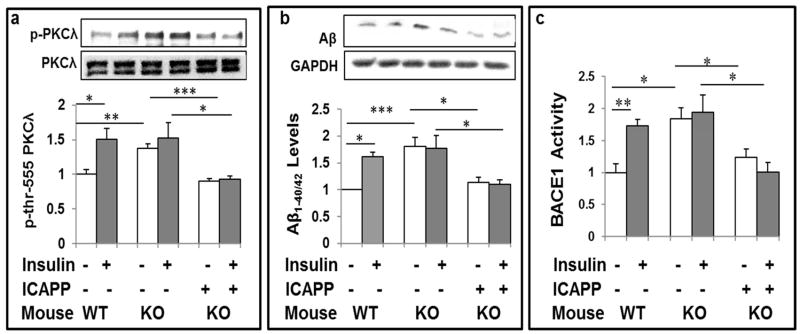
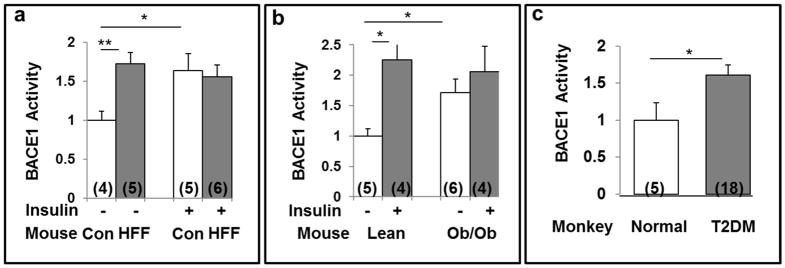
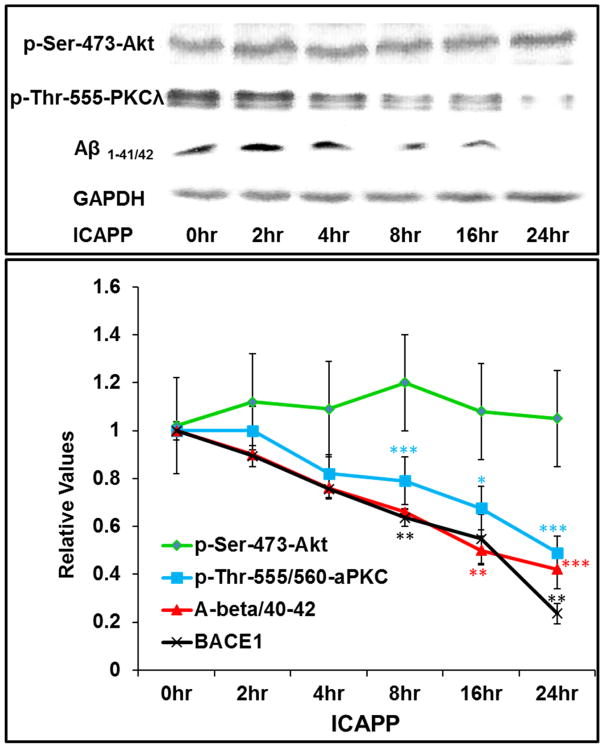
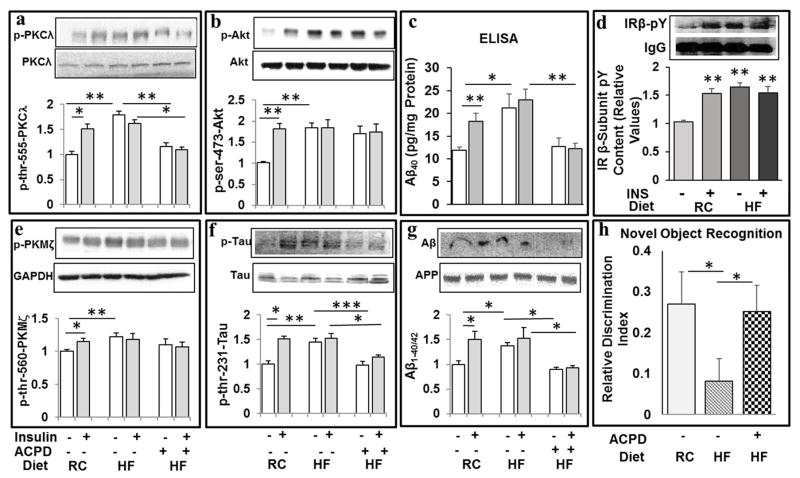
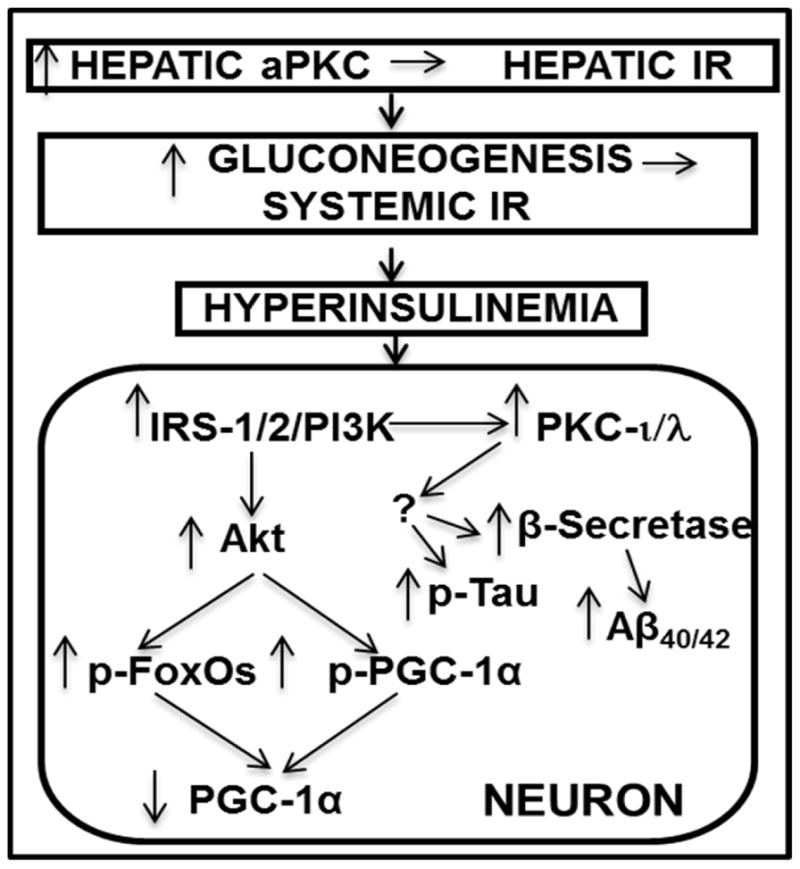
Hyperinsulinemia activates brain Akt and PKC-λ/ι and increases Aβ1-40/42 and phospho-tau in insulin-resistant animals. Here, we examined underlying mechanisms in mice, neuronal cells, and mouse hippocampal slices. Like Aβ1-40/42, β-secretase activity was increased in insulin-resistant mice and monkeys. In insulin-resistant mice, inhibition of hepatic PKC-λ/ι sufficient to correct hepatic abnormalities and hyperinsulinemia simultaneously reversed increases in Akt, atypical protein kinase C (aPKC), β-secretase, and Aβ1-40/42, and restored acute Akt activation. However, 2 aPKC inhibitors additionally blocked insulin's ability to activate brain PKC-λ/ι and thereby increase β-secretase and Aβ1-40/42. Furthermore, direct blockade of brain aPKC simultaneously corrected an impairment in novel object recognition in high-fat-fed insulin-resistant mice. In neuronal cells and/or mouse hippocampal slices, PKC-ι/λ activation by insulin, metformin, or expression of constitutive PKC-ι provoked increases in β-secretase, Aβ1-40/42, and phospho-thr-231-tau that were blocked by various PKC-λ/ι inhibitors, but not by an Akt inhibitor. PKC-λ/ι provokes increases in brain β-secretase, Aβ1-40/42, and phospho-thr-231-tau. Excessive signaling via PKC-λ/ι may link hyperinsulinemia and other PKC-λ/ι activators to pathological and functional abnormalities in Alzheimer's disease.
Keywords: Akt; Alzheimer's; Atypical PKC; Aβ; Beta-secretase; Insulin; Metformin; PKC-iota/lambda; PKM-zeta; Phospho-tau.
Published by Elsevier Inc.
Conflict of interest statement
The authors report no conflicts of interest
There are no actual or potential conflicts of interest amongst the authors.
Figures
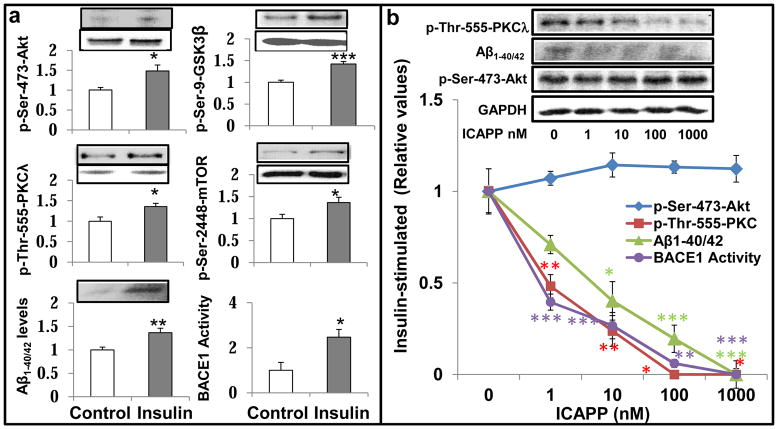
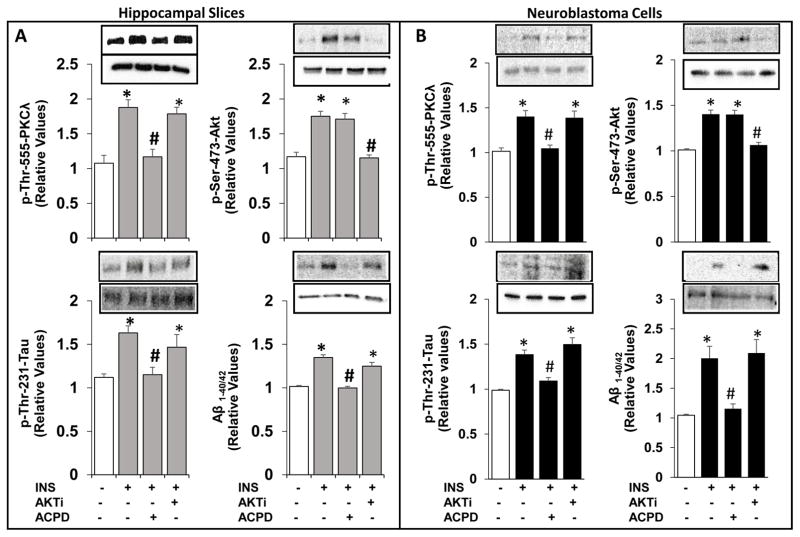
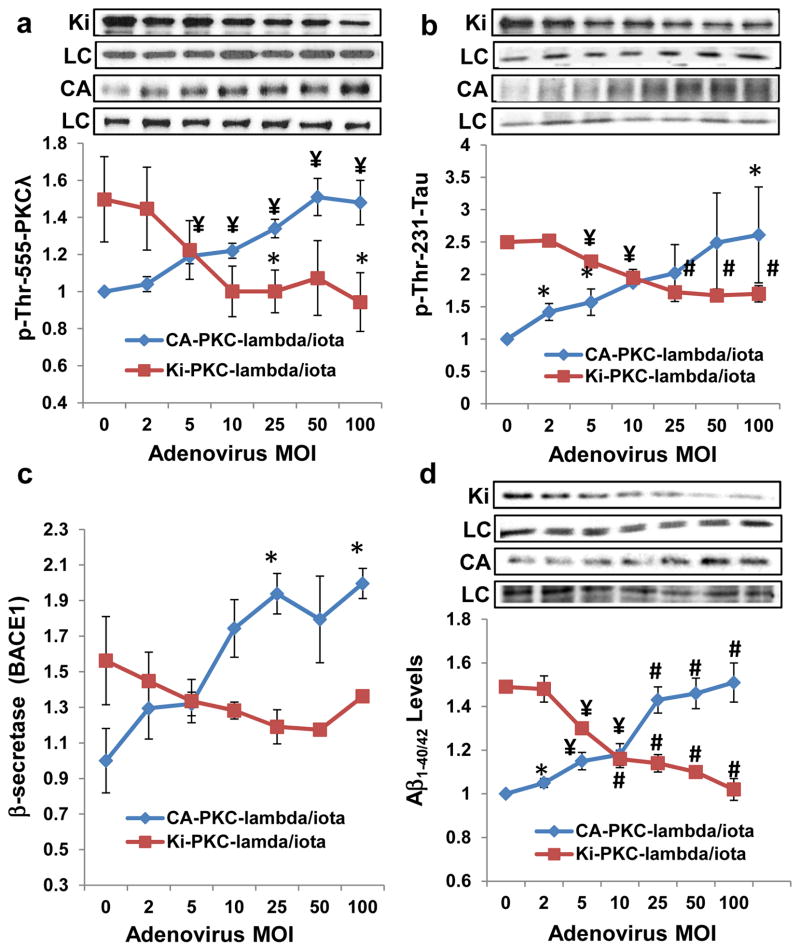
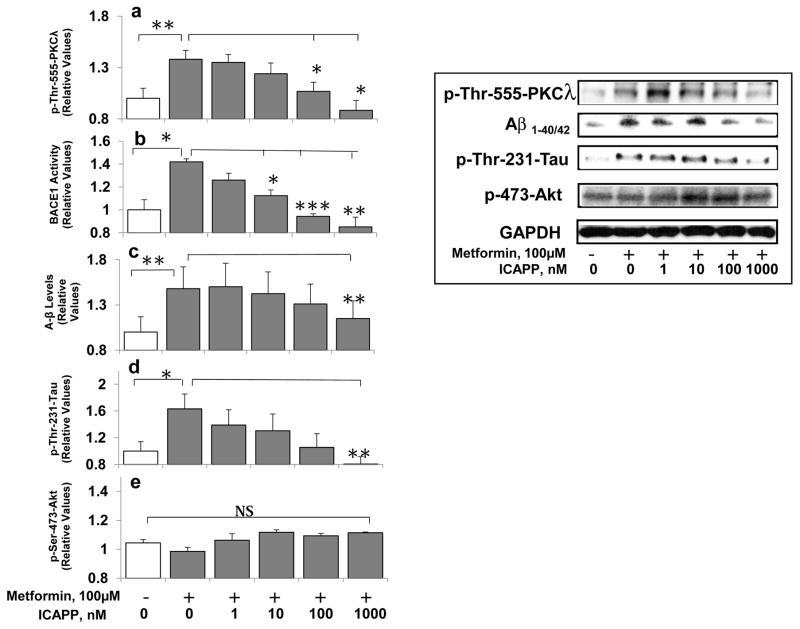
References
-
- Chen Y, Zhou K, Wang R, Liu Y, Kwak Y-D, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Luo Z, Xu H, Liao F-F. Antidiabetic drug metformin (Glucophage) increases biogenesis of Alzheimer’s amyloid peptides via upregulating BACE1 transcription. Proc Natl Acad Sci USA. 2009;106:3907–3912. - PMC - PubMed
-
- Craft S. Insulin resistance and Alzheimer’s disease pathogenesis: mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4:147–152. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
