

Analyzing the Complex Regulatory Landscape of Hfq - an Integrative, Multi-Omics Approach
- PMID: 29033902
- PMCID: PMC5627042
- DOI: 10.3389/fmicb.2017.01784
Analyzing the Complex Regulatory Landscape of Hfq - an Integrative, Multi-Omics Approach
Abstract
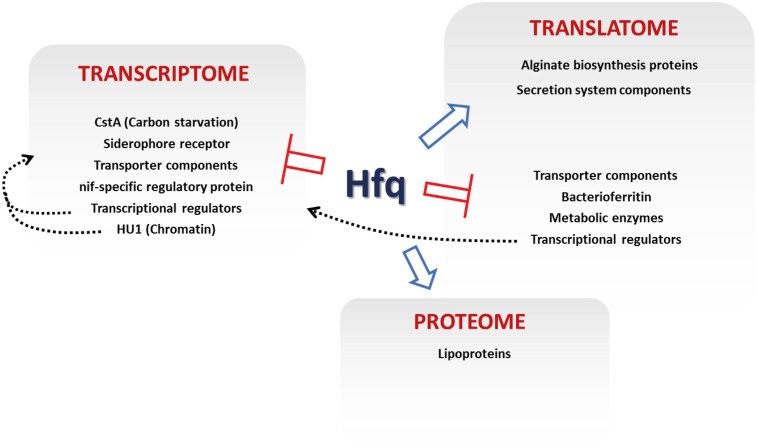
The ability of bacteria to respond to environmental change is based on the ability to coordinate, redirect and fine-tune their genetic repertoire as and when required. While we can learn a great deal from reductive analysis of individual pathways and global approaches to gene regulation, a deeper understanding of these complex signaling networks requires the simultaneous consideration of several regulatory layers at the genome scale. To highlight the power of this approach we analyzed the Hfq transcriptional/translational regulatory network in the model bacterium Pseudomonas fluorescens. We first used extensive 'omics' analyses to assess how hfq deletion affects mRNA abundance, mRNA translation and protein abundance. The subsequent, multi-level integration of these datasets allows us to highlight the discrete contributions by Hfq to gene regulation at different levels. The integrative approach to regulatory analysis we describe here has significant potential, for both dissecting individual signaling pathways and understanding the strategies bacteria use to cope with external challenges.
Keywords: Hfq; Pseudomonas; integrative approach; multi-omics analysis; ribosomal profiling.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases