

Clinical phenotype of ASD-associated DYRK1A haploinsufficiency
- PMID: 29034068
- PMCID: PMC5629761
- DOI: 10.1186/s13229-017-0173-5
Clinical phenotype of ASD-associated DYRK1A haploinsufficiency
Abstract
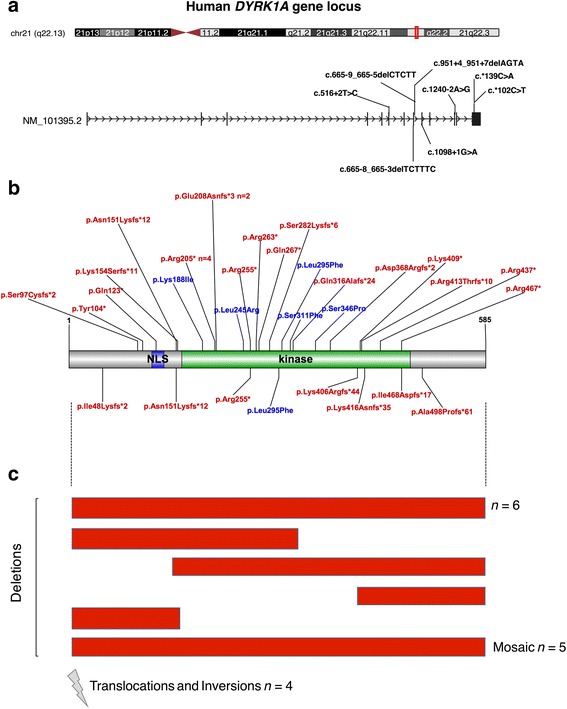
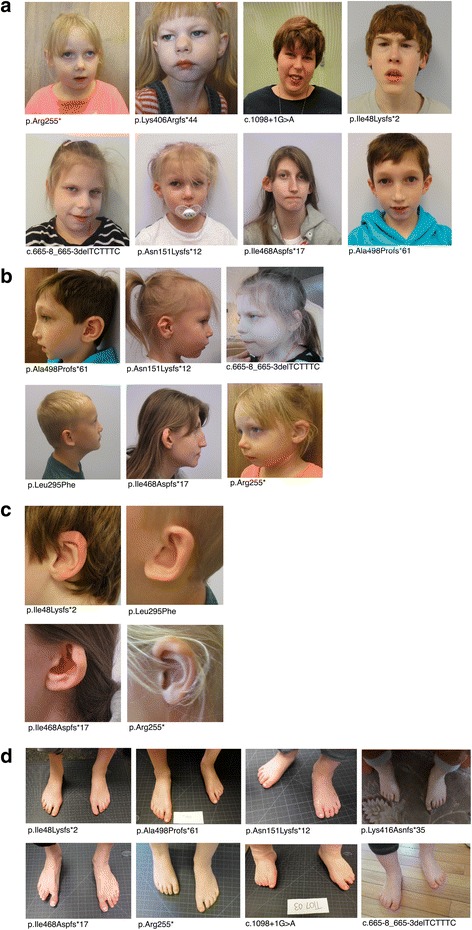
Background: DYRK1A is a gene recurrently disrupted in 0.1-0.5% of the ASD population. A growing number of case reports with DYRK1A haploinsufficiency exhibit common phenotypic features including microcephaly, intellectual disability, speech delay, and facial dysmorphisms.
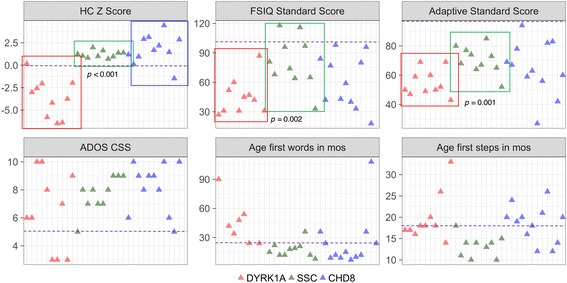
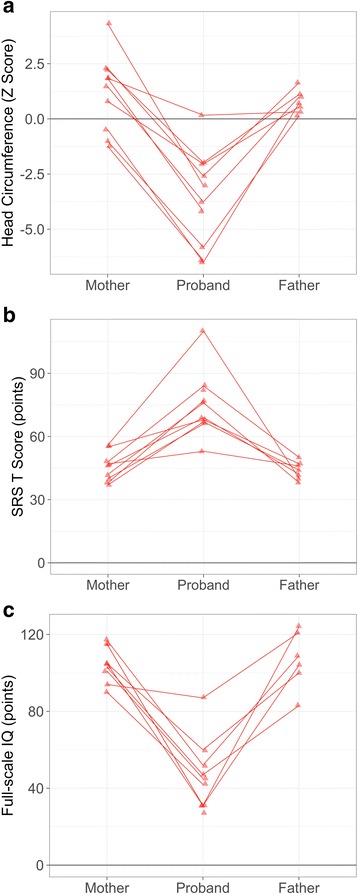
Methods: Phenotypic information from previously published DYRK1A cases (n = 51) and participants in an ongoing study at the University of Washington (UW, n = 10) were compiled. Frequencies of recurrent phenotypic features in this population were compared to features observed in a large sample with idiopathic ASD from the Simons Simplex Collection (n = 1981). UW DYRK1A cases were further characterized quantitatively and compared to a randomly subsampled set of idiopathic ASD cases matched on age and gender (n = 10) and to cases with an ASD-associated disruptive mutation to CHD8 (n = 12). Contribution of familial genetic background to clinical heterogeneity was assessed by comparing head circumference, IQ, and ASD-related symptoms of UW DYRK1A cases to their unaffected parents.
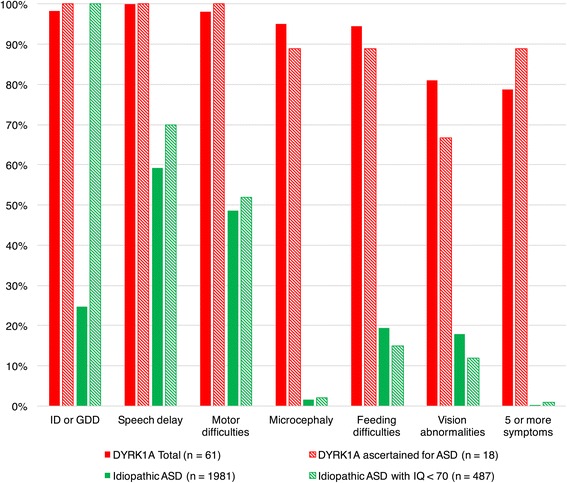
Results: DYRK1A haploinsufficiency results in a common phenotypic profile including intellectual disability, speech and motor difficulties, microcephaly, feeding difficulties, and vision abnormalities. Eighty-nine percent of DYRK1A cases ascertained for ASD presented with a constellation of five or more of these symptoms. When compared quantitatively, DYRK1A cases presented with significantly lower IQ and adaptive functioning compared to idiopathic cases and significantly smaller head size compared to both idiopathic and CHD8 cases. Phenotypic variability in parental head circumference, IQ, and ASD-related symptoms corresponded to observed variability in affected child phenotype.
Conclusions: Results confirm a core clinical phenotype for DYRK1A disruptions, with a combination of features that is distinct from idiopathic ASD. Cases with DYRK1A mutations are also distinguishable from disruptive mutations to CHD8 by head size. Measurable, quantitative characterization of DYRK1A haploinsufficiency illuminates clinical variability, which may be, in part, due to familial genetic background.
Keywords: Autism; Clinical phenotype; DYRK1A; Disruptive mutation; Genetic syndrome; Genetically defined subtype.
Conflict of interest statement
Ethics approval and consent to participate
Written consent was obtained from participants, and all procedures were approved by the University of Washington Institutional Review Board.
Consent for publication
Written informed consent was obtained from the participants for publication of their individual details and accompanying images in this manuscript. The consent form is held by the authors and is available for review by the Editor-in-Chief.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Higdon R, Earl RK, Stanberry L, Hudac CM, Montague E, Stewart E, et al. The promise of multi-omics and clinical data integration to identify and target personalized healthcare approaches in autism spectrum disorders. Omics : a journal of integrative biology. 2015;19(4):197–208. doi: 10.1089/omi.2015.0020. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
