

Coagulofibrinolytic Changes in Patients with Post-cardiac Arrest Syndrome
- PMID: 29034235
- PMCID: PMC5626829
- DOI: 10.3389/fmed.2017.00156
Coagulofibrinolytic Changes in Patients with Post-cardiac Arrest Syndrome
Abstract
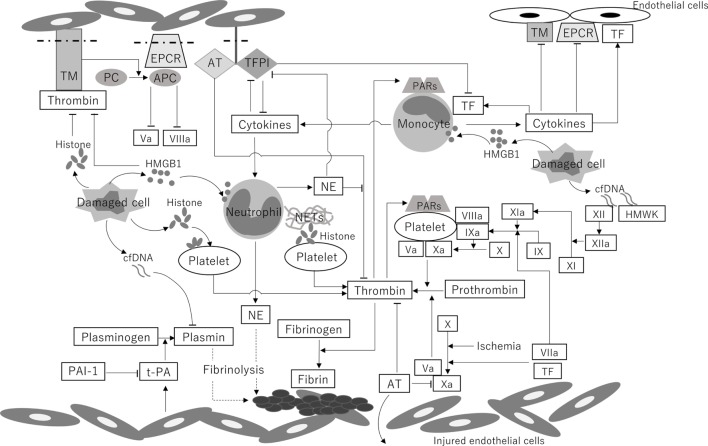
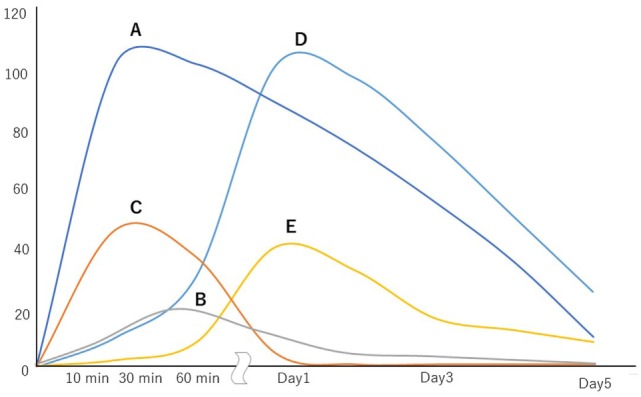
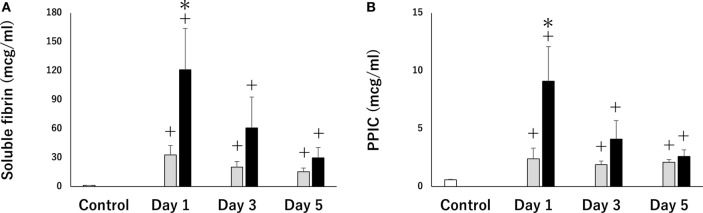
Whole-body ischemia and reperfusion due to cardiac arrest and subsequent return of spontaneous circulation constitute post-cardiac arrest syndrome (PCAS), which consists of four syndromes including systemic ischemia/reperfusion responses and post-cardiac arrest brain injury. The major pathophysiologies underlying systemic ischemia/reperfusion responses are systemic inflammatory response syndrome and increased coagulation, leading to disseminated intravascular coagulation (DIC), which clinically manifests as obstruction of microcirculation and multiple organ dysfunction. In particular, thrombotic occlusion in the brain due to DIC, referred to as the "no-reflow phenomenon," may be deeply involved in post-cardiac arrest brain injury, which is the leading cause of mortality in patients with PCAS. Coagulofibrinolytic changes in patients with PCAS are characterized by tissue factor-dependent coagulation, which is accelerated by impaired anticoagulant mechanisms, including antithrombin, protein C, thrombomodulin, and tissue factor pathway inhibitor. Damage-associated molecular patterns (DAMPs) accelerate not only tissue factor-dependent coagulation but also the factor XII- and factor XI-dependent activation of coagulation. Inflammatory cytokines are also involved in these changes via the expression of tissue factor on endothelial cells and monocytes, the inhibition of anticoagulant systems, and the release of neutrophil elastase from neutrophils activated by inflammatory cytokines. Hyperfibrinolysis in the early phase of PCAS is followed by inadequate endogenous fibrinolysis and fibrinolytic shutdown by plasminogen activator inhibitor-1. Moreover, cell-free DNA, which is also a DAMP, plays a pivotal role in the inhibition of fibrinolysis. DIC diagnosis criteria or fibrinolysis markers, including d-dimer and fibrin/fibrinogen degradation products, which are commonly tested in patients and easily accessible, can be used to predict the mortality or neurological outcome of PCAS patients with high accuracy. A number of studies have explored therapy for this unique pathophysiology since the first report on "no-reflow phenomenon" was published roughly 50 years ago. However, the optimum therapeutic strategy focusing on the coagulofibrinolytic changes in cardiac arrest or PCAS patients has not yet been established. The elucidation of more precise pathomechanisms of coagulofibrinolytic changes in PCAS may aid in the development of novel therapeutic targets, leading to an improvement in the outcomes of PCAS patients.
Keywords: activation of coagulation; disseminated intravascular coagulation; fibrinolytic shutdown; impaired anticoagulant; no-reflow phenomenon; post-cardiac arrest syndrome; systemic ischemia/reperfusion.
Figures
Similar articles
-
Association of Histones With Coagulofibrinolytic Responses and Organ Dysfunction in Adult Post-cardiac Arrest Syndrome.Front Cardiovasc Med. 2022 Jun 28;9:885406. doi: 10.3389/fcvm.2022.885406. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 35837604 Free PMC article.
-
Coagulofibrinolytic changes in patients with disseminated intravascular coagulation associated with post-cardiac arrest syndrome--fibrinolytic shutdown and insufficient activation of fibrinolysis lead to organ dysfunction.Thromb Res. 2013 Jul;132(1):e64-9. doi: 10.1016/j.thromres.2013.05.010. Epub 2013 May 30. Thromb Res. 2013. PMID: 23726093
-
Disseminated intravascular coagulation in cardiac arrest and resuscitation.J Thromb Haemost. 2019 Aug;17(8):1205-1216. doi: 10.1111/jth.14480. Epub 2019 Jun 5. J Thromb Haemost. 2019. PMID: 31102491 Review.
-
The activation of neutrophil elastase-mediated fibrinolysis is not sufficient to overcome the fibrinolytic shutdown of disseminated intravascular coagulation associated with systemic inflammation.Thromb Res. 2007;121(1):67-73. doi: 10.1016/j.thromres.2007.02.010. Epub 2007 Mar 29. Thromb Res. 2007. PMID: 17397908
-
Disseminated intravascular coagulation in trauma patients.Semin Thromb Hemost. 2001 Dec;27(6):585-92. doi: 10.1055/s-2001-18864. Semin Thromb Hemost. 2001. PMID: 11740682 Review.
Cited by
-
Coagulofibrinolytic effects of recombinant soluble thrombomodulin in prolonged porcine cardiac arrest.Resusc Plus. 2023 Sep 27;16:100477. doi: 10.1016/j.resplu.2023.100477. eCollection 2023 Dec. Resusc Plus. 2023. PMID: 37811363 Free PMC article.
-
Disseminated intravascular coagulation is associated with a poor outcome in patients with out-of-hospital cardiac arrest receiving VA-ECMO.J Artif Organs. 2025 Jan 6. doi: 10.1007/s10047-024-01487-3. Online ahead of print. J Artif Organs. 2025. PMID: 39760969
-
Effect of cardioplegic arrest and reperfusion on left and right ventricular proteome/phosphoproteome in patients undergoing surgery for coronary or aortic valve disease.Int J Mol Med. 2022 Jun;49(6):77. doi: 10.3892/ijmm.2022.5133. Epub 2022 Apr 15. Int J Mol Med. 2022. PMID: 35425992 Free PMC article.
-
Coagulopathy Induced by Veno-Arterial Extracorporeal Membrane Oxygenation Is Associated With a Poor Outcome in Patients With Out-of-Hospital Cardiac Arrest.Front Med (Lausanne). 2021 Apr 30;8:651832. doi: 10.3389/fmed.2021.651832. eCollection 2021. Front Med (Lausanne). 2021. PMID: 34017845 Free PMC article.
-
Association of Histones With Coagulofibrinolytic Responses and Organ Dysfunction in Adult Post-cardiac Arrest Syndrome.Front Cardiovasc Med. 2022 Jun 28;9:885406. doi: 10.3389/fcvm.2022.885406. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 35837604 Free PMC article.
References
-
- Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Böttiger BW, et al. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation (2008) 118:2452–83.10.1161/CIRCULATIONAHA.108.190652 - DOI - PubMed
-
- Adrie C, Adib-Conquy M, Laurent I, Monchi M, Vinsonneau C, Fitting C, et al. Successful cardiopulmonary resuscitation after cardiac arrest as a “sepsis-like” syndrome. Circulation (2002) 106:562–8.10.1161/01.CIR.0000023891.80661.AD - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources