

N6-adenine DNA methylation is associated with the linker DNA of H2A.Z-containing well-positioned nucleosomes in Pol II-transcribed genes in Tetrahymena
- PMID: 29036602
- PMCID: PMC5714169
- DOI: 10.1093/nar/gkx883
N6-adenine DNA methylation is associated with the linker DNA of H2A.Z-containing well-positioned nucleosomes in Pol II-transcribed genes in Tetrahymena
Abstract
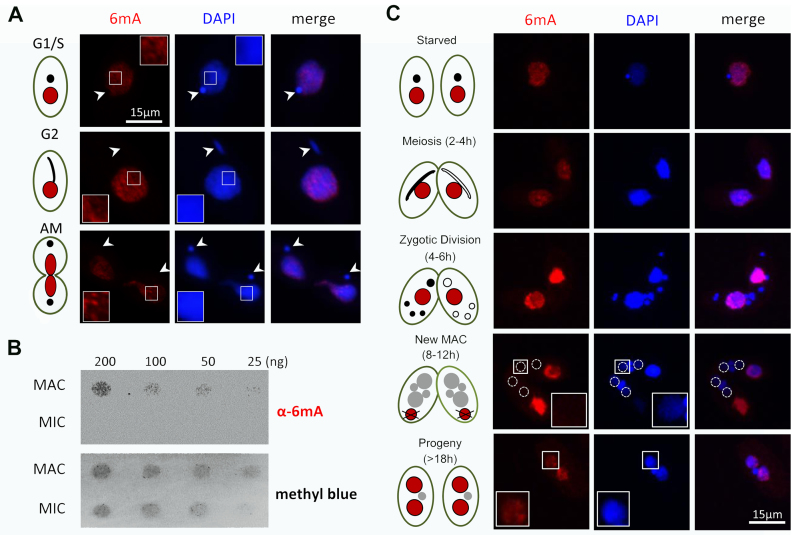
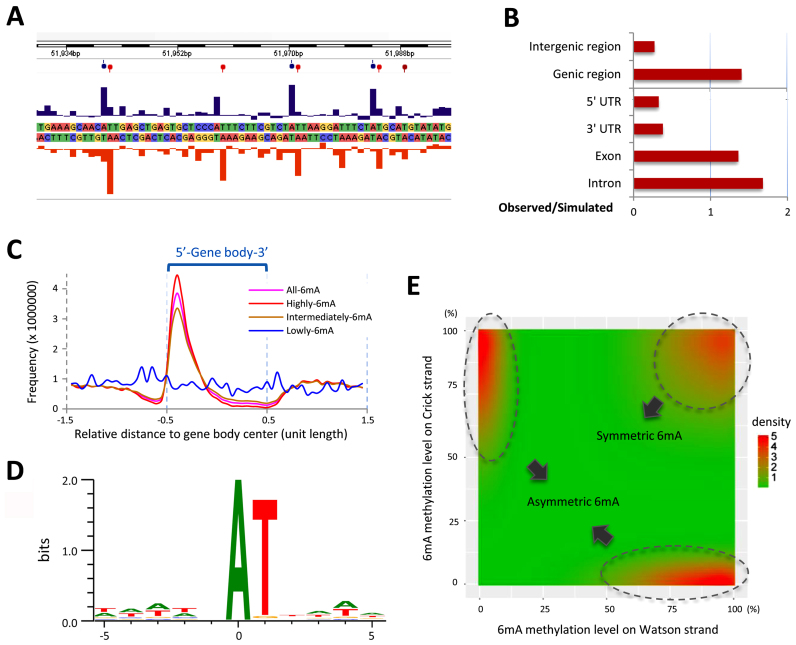
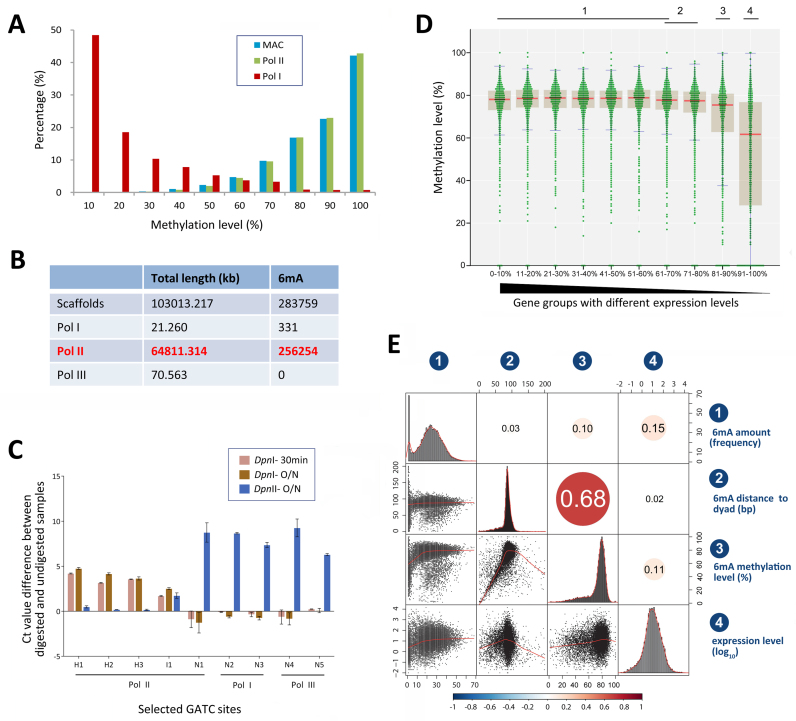
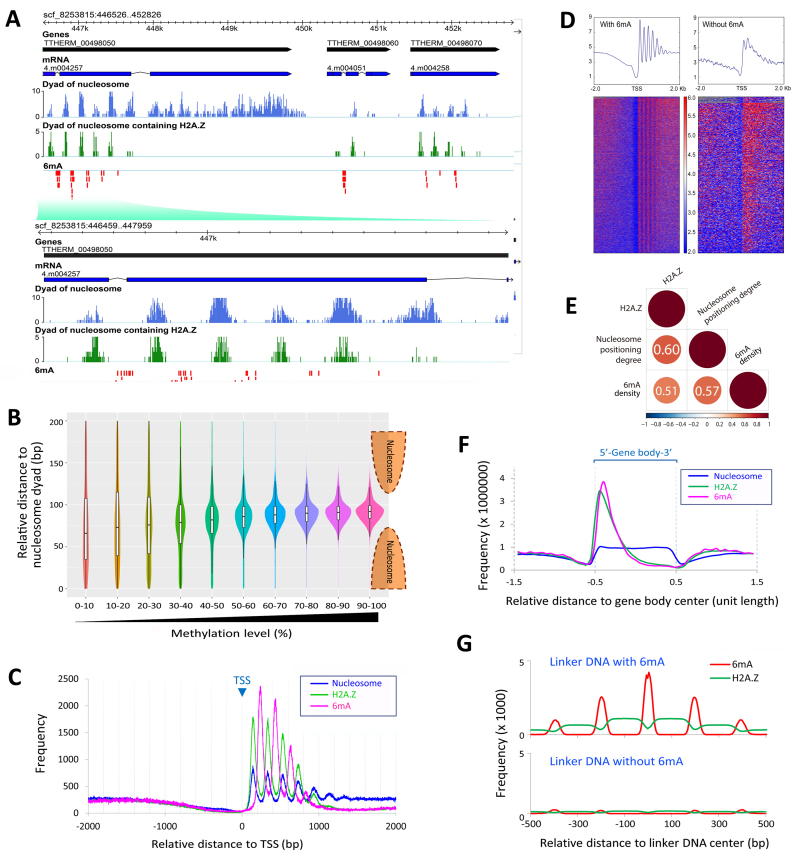
DNA N6-methyladenine (6mA) is newly rediscovered as a potential epigenetic mark across a more diverse range of eukaryotes than previously realized. As a unicellular model organism, Tetrahymena thermophila is among the first eukaryotes reported to contain 6mA modification. However, lack of comprehensive information about 6mA distribution hinders further investigations into its function and regulatory mechanism. In this study, we provide the first genome-wide, base pair-resolution map of 6mA in Tetrahymena by applying single-molecule real-time (SMRT) sequencing. We provide evidence that 6mA occurs mostly in the AT motif of the linker DNA regions. More strikingly, these linker DNA regions with 6mA are usually flanked by well-positioned nucleosomes and/or H2A.Z-containing nucleosomes. We also find that 6mA is exclusively associated with RNA polymerase II (Pol II)-transcribed genes, but is not an unambiguous mark for active transcription. These results support that 6mA is an integral part of the chromatin landscape shaped by adenosine triphosphate (ATP)-dependent chromatin remodeling and transcription.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
