

Mitochondria and Mitochondrial Cascades in Alzheimer's Disease
- PMID: 29036828
- PMCID: PMC5869994
- DOI: 10.3233/JAD-170585
Mitochondria and Mitochondrial Cascades in Alzheimer's Disease
Abstract
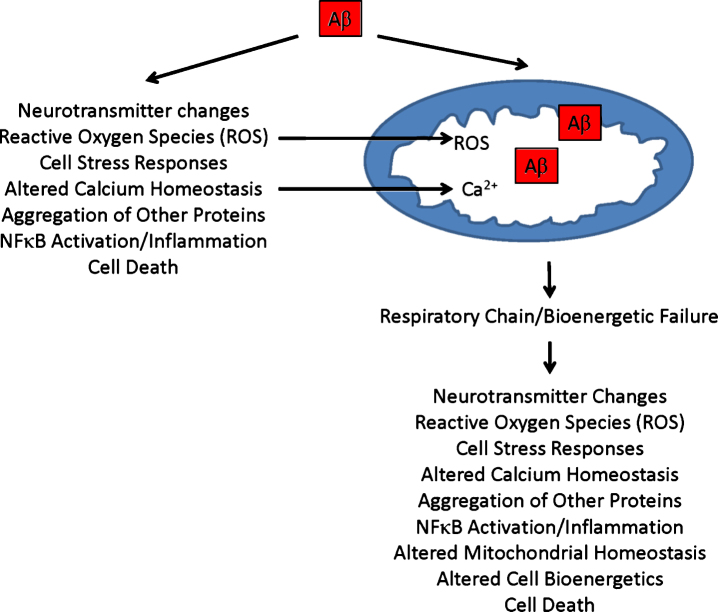
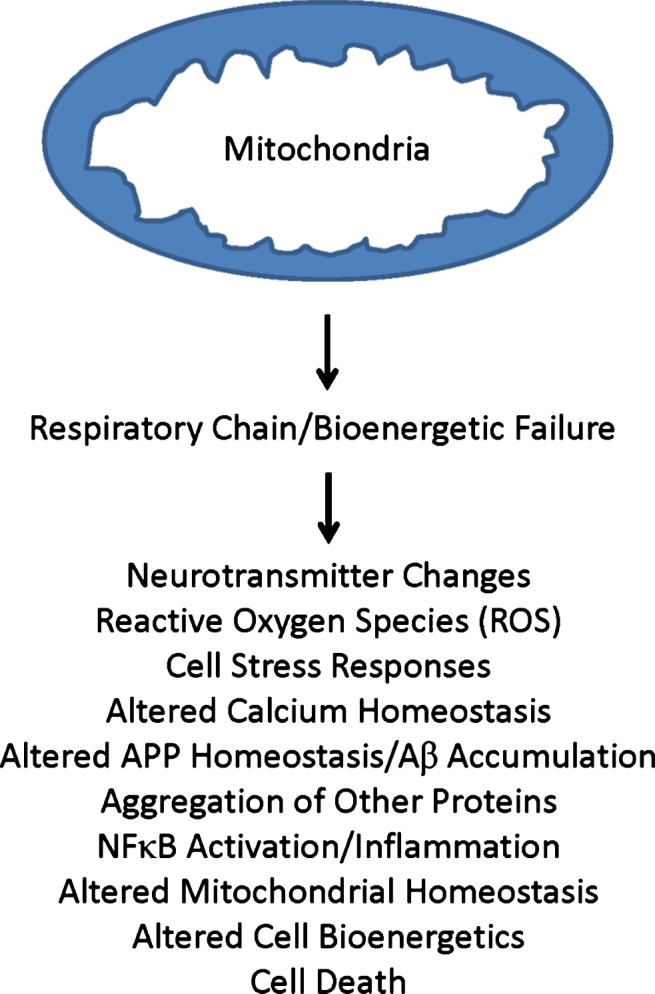
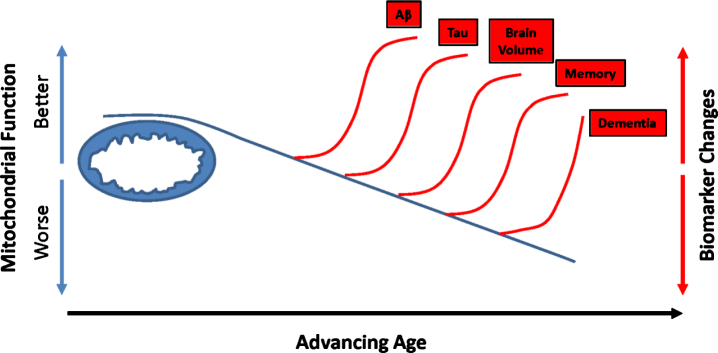
Decades of research indicate mitochondria from Alzheimer's disease (AD) patients differ from those of non-AD individuals. Initial studies revealed structural differences, and subsequent studies showed functional deficits. Observations of structure and function changes prompted investigators to consider the consequences, significance, and causes of AD-related mitochondrial dysfunction. Currently, extensive research argues mitochondria may mediate, drive, or contribute to a variety of AD pathologies. The perceived significance of these mitochondrial changes continues to grow, and many currently believe AD mitochondrial dysfunction represents a reasonable therapeutic target. Debate continues over the origin of AD mitochondrial changes. Some argue amyloid-β (Aβ) induces AD mitochondrial dysfunction, a view that does not challenge the amyloid cascade hypothesis and that may in fact help explain that hypothesis. Alternatively, data indicate mitochondrial dysfunction exists independent of Aβ, potentially lies upstream of Aβ deposition, and suggest a primary mitochondrial cascade hypothesis that assumes mitochondrial pathology hierarchically supersedes Aβ pathology. Mitochondria, therefore, appear at least to mediate or possibly even initiate pathologic molecular cascades in AD. This review considers studies and data that inform this area of AD research.
Keywords: Alzheimer’s disease; bioenergetics; cascade; cytochrome oxidase; mitochondria.
Figures
References
-
- Johnson AB, Blum NR (1970) Nucleoside phosphatase activities associated with the tangles and plaques of Alzheimer’s disease: A histochemical study of natural and experimental neurofibrillary tangles. J Neuropathol Exp Neurol 29, 463–478. - PubMed
-
- Wisniewski H, Terry RD, Hirano A (1970) Neurofibrillary pathology. J Neuropathol Exp Neurol 29, 163–176. - PubMed
-
- Baloyannis SJ (2006) Mitochondrial alterations in Alzheimer’s disease. J Alzheimers Dis 9, 119–126. - PubMed
-
- Ferris SH, de Leon MJ, Wolf AP, Farkas T, Christman DR, Reisberg B, Fowler JS, Macgregor R, Goldman A, George AE, Rampal S (1980) Positron emission tomography in the study of aging and senile dementia. Neurobiol Aging 1, 127–131. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
