

Neuropathological Comparison of Adult Onset and Juvenile Huntington's Disease with Cerebellar Atrophy: A Report of a Father and Son
- PMID: 29036832
- PMCID: PMC5832043
- DOI: 10.3233/JHD-170261
Neuropathological Comparison of Adult Onset and Juvenile Huntington's Disease with Cerebellar Atrophy: A Report of a Father and Son
Abstract
Background: Huntington's disease (HD) is an autosomal dominant neurodegenerative disease caused by a trinucleotide (CAG) repeat expansion in huntingtin (HTT) on chromosome 4. Anticipation can cause longer repeat expansions in children of HD patients. Juvenile Huntington's disease (JHD), defined as HD arising before age 20, accounts for 5-10% of HD cases, with cases arising in the first decade accounting for approximately 1%. Clinically, JHD differs from the predominately choreiform adult onset Huntington's disease (AOHD) with variable presentations, including symptoms such as myoclonus, seizures, Parkinsonism, and cognitive decline.
Objective: The neuropathologic changes of AOHD are well characterized, but there are fewer reports that describe the neuropathology of JHD. Here we report a case of a six-year-old boy with paternally-inherited JHD caused by 169 CAG trinucleotide repeats who presented at age four with developmental delay, dysarthria, and seizures before dying at age 6. The boy's clinical presentation and neuropathological findings are directly compared to those of his father, who presented with AOHD and 54 repeats.
Methods: A full autopsy was performed for the JHD case and a brain-only autopsy was performed for the AOHD case. Histochemically- and immunohistochemically-stained slides were prepared from formalin-fixed, paraffin-embedded tissue sections.
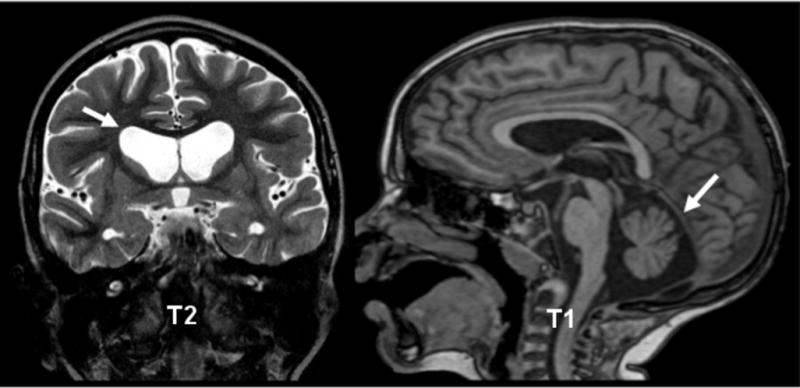
Results: Both cases had neuropathology corresponding to Vonsattel grade 3. The boy also had cerebellar atrophy with huntingtin-positive inclusions in the cerebellum, findings not present in the father.
Conclusions: Autopsies of father and son provide a unique opportunity to compare and contrast the neuropathologic findings of juvenile and adult onset HD while also providing the first immunohistochemical evidence of cerebellar involvement in JHD. Additionally this is the first known report to include findings from peripheral tissue in a case of JHD.
Keywords: Autopsy; Huntington’s disease; huntingtin protein; immunohistochemistry; juvenile Huntington’s disease; literature review; neuropathology.
Conflict of interest statement
Figures




Similar articles
-
Juvenile Huntington disease in an 18-month-old boy revealed by global developmental delay and reduced cerebellar volume.Am J Med Genet A. 2011 Apr;155A(4):815-8. doi: 10.1002/ajmg.a.33911. Epub 2011 Mar 15. Am J Med Genet A. 2011. PMID: 21412977
-
Dysregulation of Human Juvenile Huntington's Disease Brain Proteomes in Cortex and Putamen Involves Mitochondrial and Neuropeptide Systems.J Huntingtons Dis. 2023;12(4):315-333. doi: 10.3233/JHD-230577. J Huntingtons Dis. 2023. PMID: 38108356
-
Psychiatric and cognitive difficulties as indicators of juvenile huntington disease onset in 29 patients.Arch Neurol. 2007 Jun;64(6):813-9. doi: 10.1001/archneur.64.6.813. Arch Neurol. 2007. PMID: 17562929
-
Tics as an initial manifestation of juvenile Huntington's disease: case report and literature review.BMC Neurol. 2017 Aug 8;17(1):152. doi: 10.1186/s12883-017-0923-1. BMC Neurol. 2017. PMID: 28789621 Free PMC article. Review.
-
Clinical Presentation and Features of Juvenile-Onset Huntington's Disease: A Systematic Review.J Huntingtons Dis. 2019;8(2):171-179. doi: 10.3233/JHD-180339. J Huntingtons Dis. 2019. PMID: 31045518
Cited by
-
Patterns of CAG repeat instability in the central nervous system and periphery in Huntington's disease and in spinocerebellar ataxia type 1.Hum Mol Genet. 2020 Aug 29;29(15):2551-2567. doi: 10.1093/hmg/ddaa139. Hum Mol Genet. 2020. PMID: 32761094 Free PMC article.
-
DRPLA: understanding the natural history and developing biomarkers to accelerate therapeutic trials in a globally rare repeat expansion disorder.J Neurol. 2021 Aug;268(8):3031-3041. doi: 10.1007/s00415-020-10218-6. Epub 2020 Oct 26. J Neurol. 2021. PMID: 33106889 Free PMC article.
-
Juvenile-Onset Huntington Disease Pathophysiology and Neurodevelopment: A Review.Mov Disord. 2022 Jan;37(1):16-24. doi: 10.1002/mds.28823. Epub 2021 Oct 12. Mov Disord. 2022. PMID: 34636452 Free PMC article. Review.
-
Brain structure in juvenile-onset Huntington disease.Neurology. 2019 Apr 23;92(17):e1939-e1947. doi: 10.1212/WNL.0000000000007355. Epub 2019 Apr 10. Neurology. 2019. PMID: 30971481 Free PMC article.
-
Molecular Mechanisms and Therapeutics for SBMA/Kennedy's Disease.Neurotherapeutics. 2019 Oct;16(4):928-947. doi: 10.1007/s13311-019-00790-9. Neurotherapeutics. 2019. PMID: 31686397 Free PMC article. Review.
References
-
- Vonsattel JP, Keller C, Cortes Ramirez EP. Huntington’s disease - neuropathology. Handbook of clinical neurology. 2011;100:83–100. - PubMed
-
- Milunsky JM, Maher TA, Loose BA, Darras BT, Ito M. XL PCR for the detection of large trinucleotide expansions in juvenile Huntington’s disease. Clinical genetics. 2003;64(1):70–3. - PubMed
-
- Cloud LJ, Rosenblatt A, Margolis RL, Ross CA, Pillai JA, Corey-Bloom J, et al. Seizures in juvenile Huntington’s disease: frequency and characterization in a multicenter cohort. Movement disorders: official journal of the Movement Disorder Society. 2012;27(14):1797–800. - PubMed
-
- Gonzalez-Alegre P, Afifi AK. Clinical characteristics of childhood-onset (juvenile) Huntington disease: report of 12 patients and review of the literature. Journal of child neurology. 2006;21(3):223–9. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
