

A High-Resolution Genome-Wide CRISPR/Cas9 Viability Screen Reveals Structural Features and Contextual Diversity of the Human Cell-Essential Proteome
- PMID: 29038160
- PMCID: PMC5730719
- DOI: 10.1128/MCB.00302-17
A High-Resolution Genome-Wide CRISPR/Cas9 Viability Screen Reveals Structural Features and Contextual Diversity of the Human Cell-Essential Proteome
Abstract
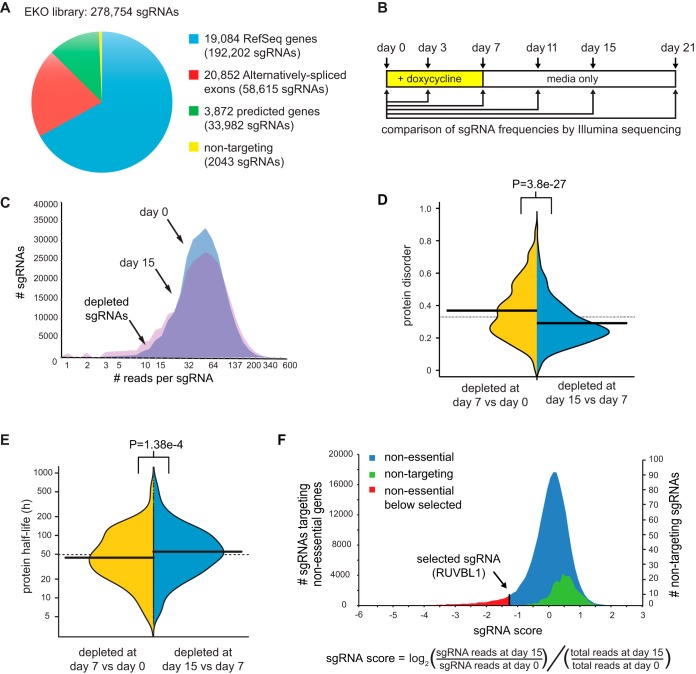
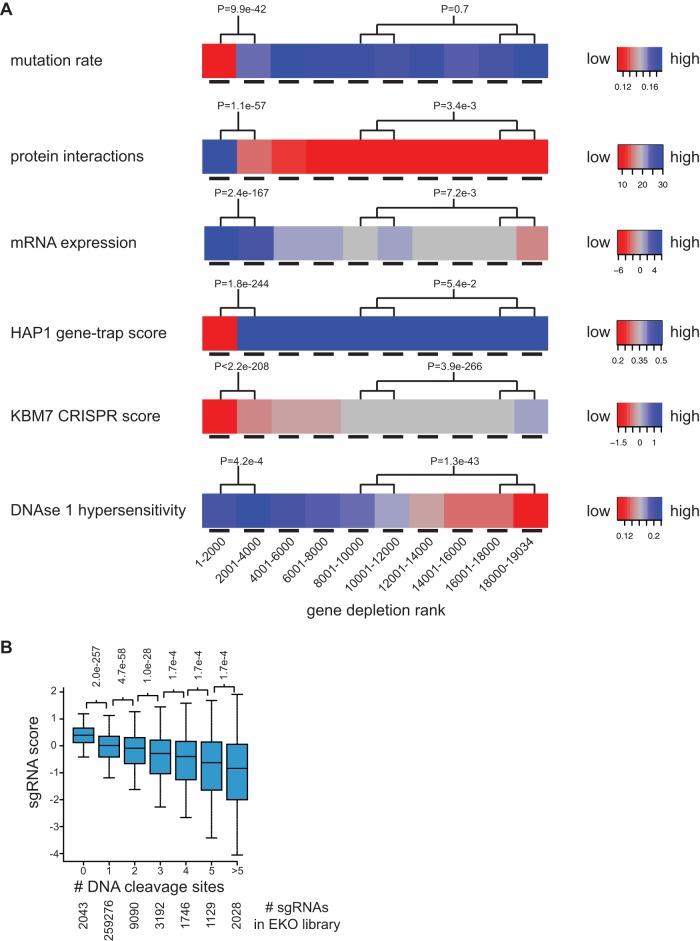
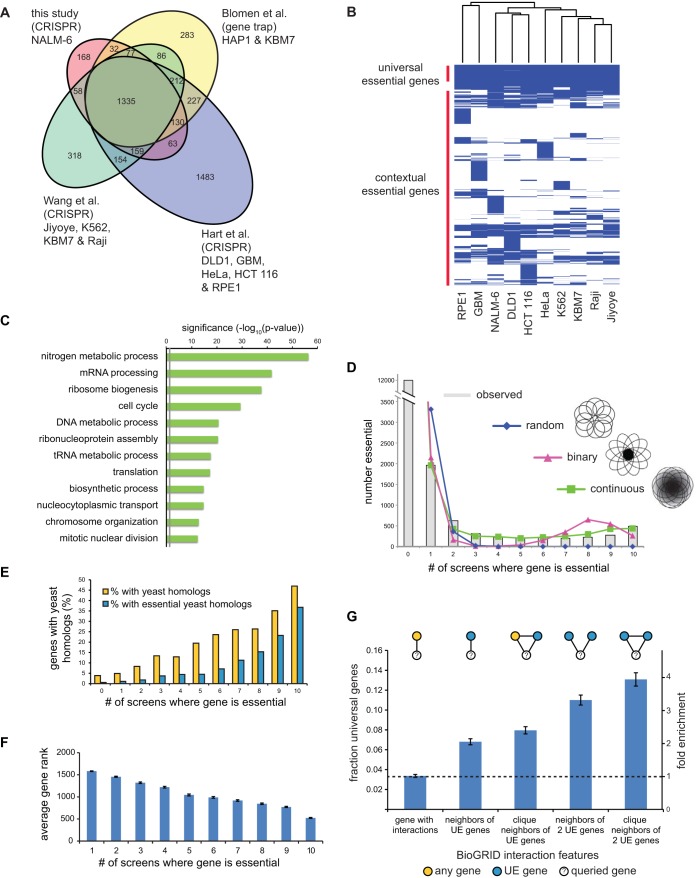
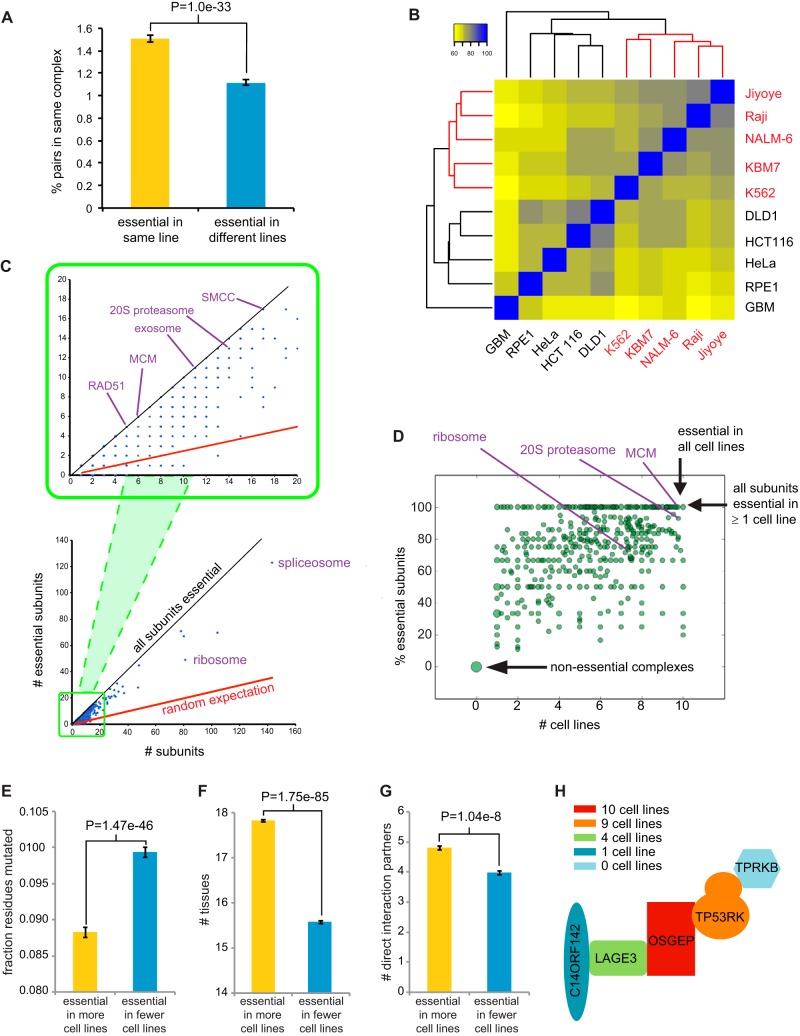
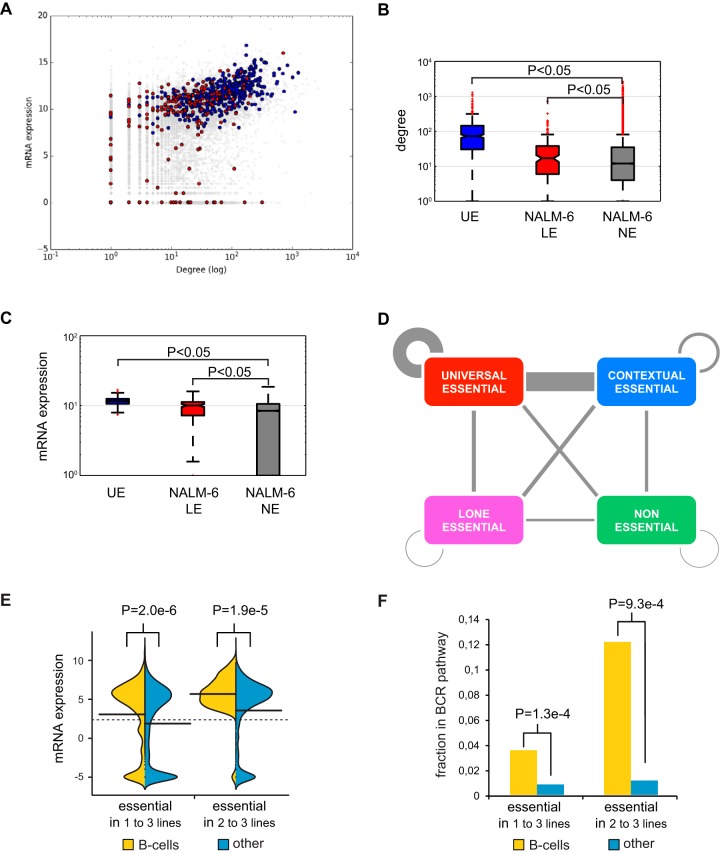
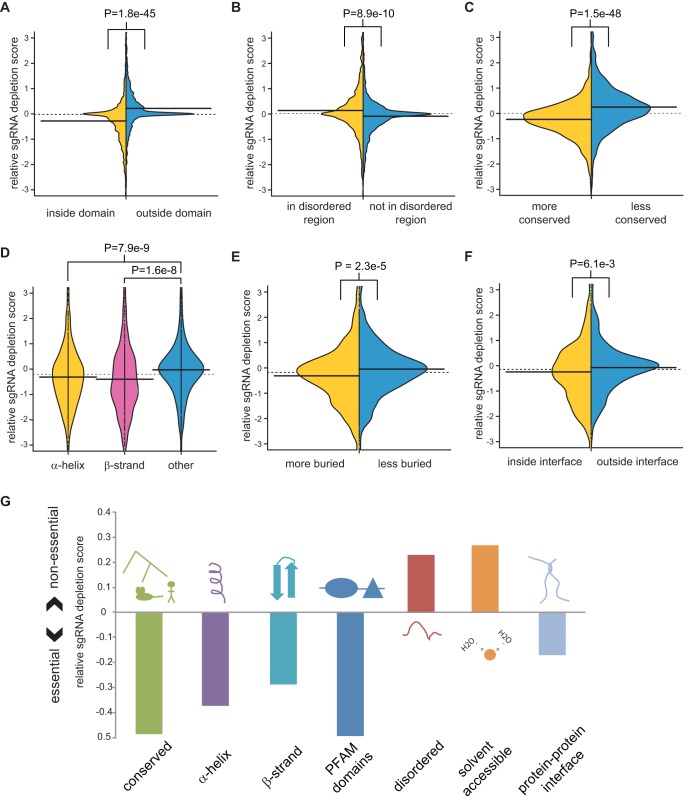
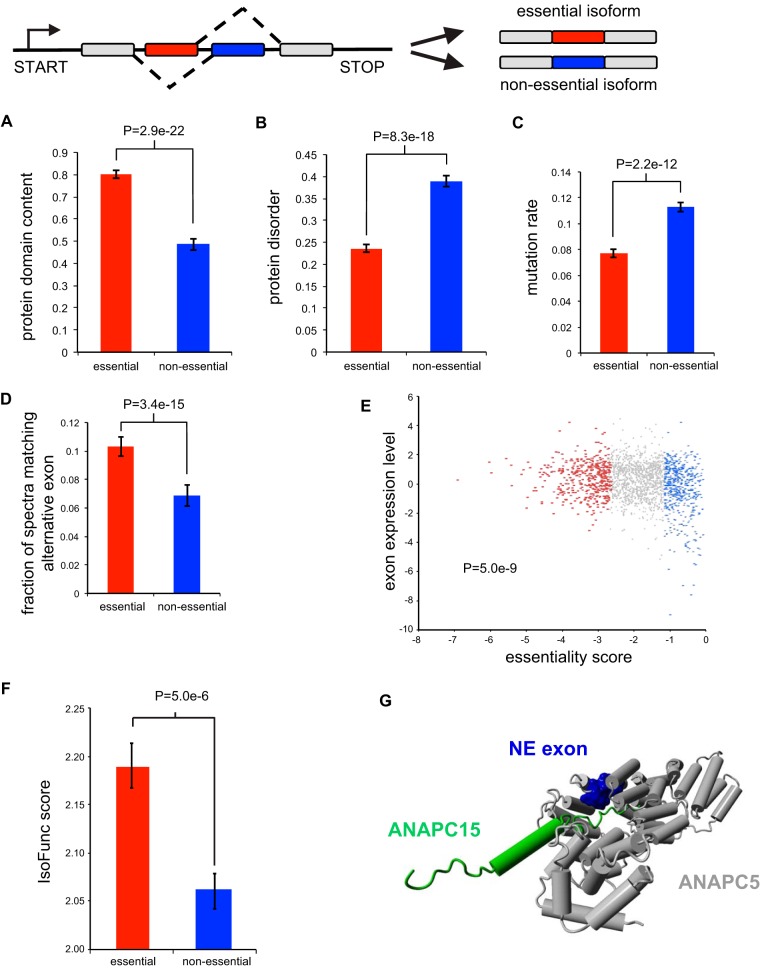
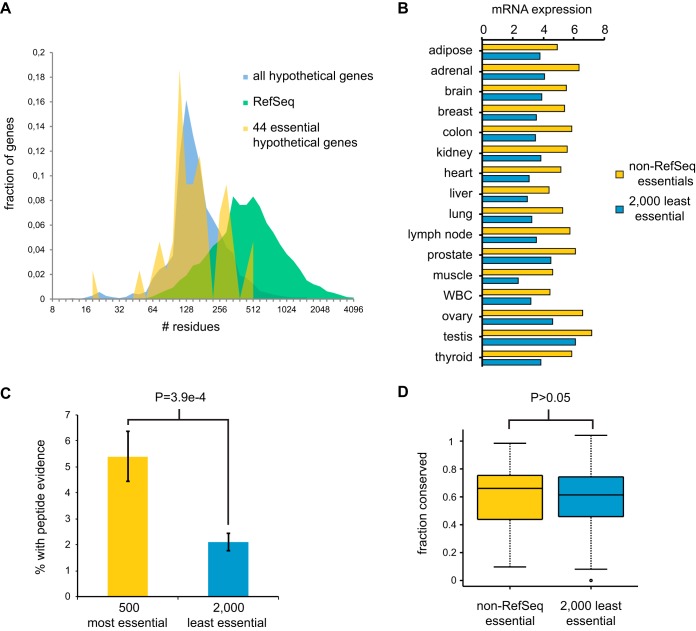
To interrogate genes essential for cell growth, proliferation and survival in human cells, we carried out a genome-wide clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 screen in a B-cell lymphoma line using a custom extended-knockout (EKO) library of 278,754 single-guide RNAs (sgRNAs) that targeted 19,084 RefSeq genes, 20,852 alternatively spliced exons, and 3,872 hypothetical genes. A new statistical analysis tool called robust analytics and normalization for knockout screens (RANKS) identified 2,280 essential genes, 234 of which were unique. Individual essential genes were validated experimentally and linked to ribosome biogenesis and stress responses. Essential genes exhibited a bimodal distribution across 10 different cell lines, consistent with a continuous variation in essentiality as a function of cell type. Genes essential in more lines had more severe fitness defects and encoded the evolutionarily conserved structural cores of protein complexes, whereas genes essential in fewer lines formed context-specific modules and encoded subunits at the periphery of essential complexes. The essentiality of individual protein residues across the proteome correlated with evolutionary conservation, structural burial, modular domains, and protein interaction interfaces. Many alternatively spliced exons in essential genes were dispensable and were enriched for disordered regions. Fitness defects were observed for 44 newly evolved hypothetical reading frames. These results illuminate the contextual nature and evolution of essential gene functions in human cells.
Keywords: CRISPR/Cas9; alternative splicing; gene essentiality; genetic screen; hypothetical gene; protein complex; proteome.
Copyright © 2017 American Society for Microbiology.
Figures
References
-
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, et al. . 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391. doi:10.1038/nature00935. - DOI - PubMed
-
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M, Deshpande R, Li Z, Lin ZY, Liang W, Marback M, Paw J, San Luis BJ, Shuteriqi E, Tong AH, van Dyk N, Wallace IM, Whitney JA, Weirauch MT, Zhong G, Zhu H, Houry WA, Brudno M, Ragibizadeh S, Papp B, Pal C, Roth FP, Giaever G, Nislow C, Troyanskaya OG, Bussey H, Bader GD, Gingras AC, Morris QD, Kim PM, Kaiser CA, et al. . 2010. The genetic landscape of a cell. Science 327:425–431. doi:10.1126/science.1180823. - DOI - PMC - PubMed
-
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, Pelechano V, Styles EB, Billmann M, van Leeuwen J, van Dyk N, Lin ZY, Kuzmin E, Nelson J, Piotrowski JS, Srikumar T, Bahr S, Chen Y, Deshpande R, Kurat CF, Li SC, Li Z, Usaj MM, Okada H, Pascoe N, San Luis BJ, Sharifpoor S, Shuteriqi E, Simpkins SW, Snider J, Suresh HG, Tan Y, Zhu H, Malod-Dognin N, Janjic V, Przulj N, Troyanskaya OG, Stagljar I, Xia T, Ohya Y, Gingras AC, Raught B, Boutros M, Steinmetz LM, Moore CL, Rosebrock AP, et al. . 2016. A global genetic interaction network maps a wiring diagram of cellular function. Science 353:aaf1420. doi:10.1126/science.aaf1420. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources