

CB1-Dependent Long-Term Depression in Ventral Tegmental Area GABA Neurons: A Novel Target for Marijuana
- PMID: 29038246
- PMCID: PMC5678024
- DOI: 10.1523/JNEUROSCI.0190-17.2017
CB1-Dependent Long-Term Depression in Ventral Tegmental Area GABA Neurons: A Novel Target for Marijuana
Abstract
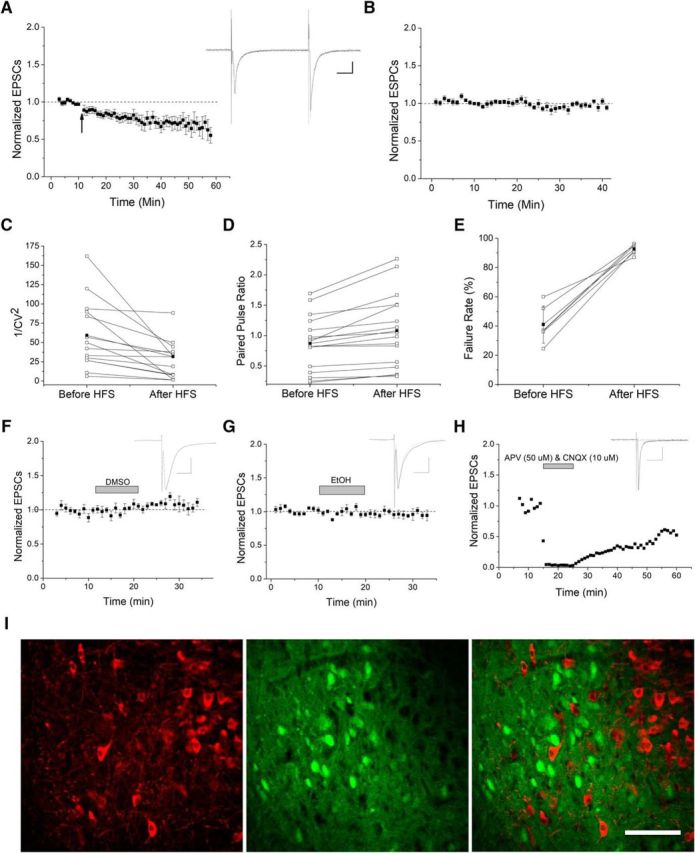
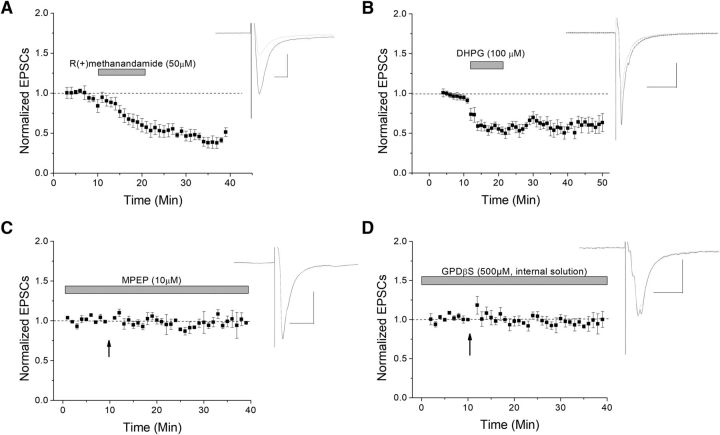
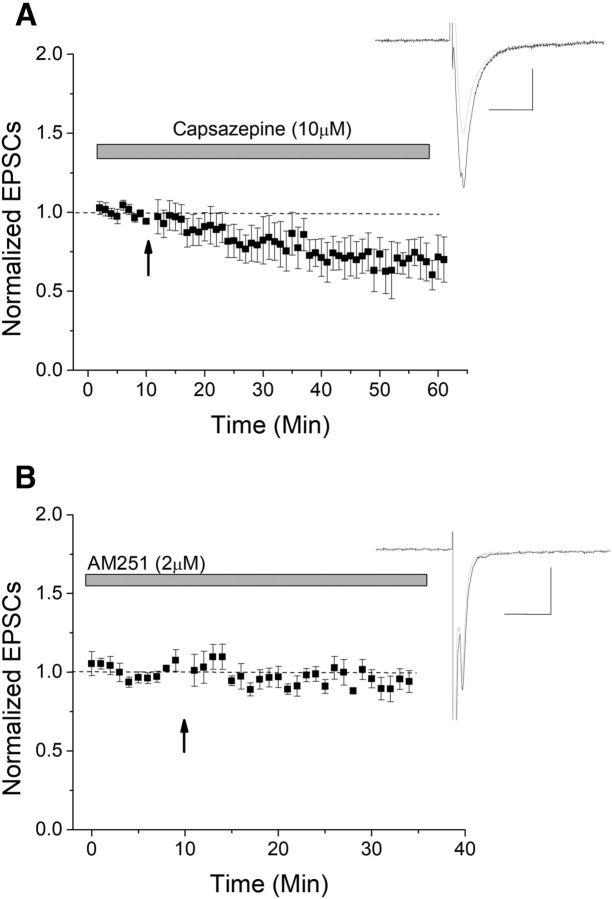
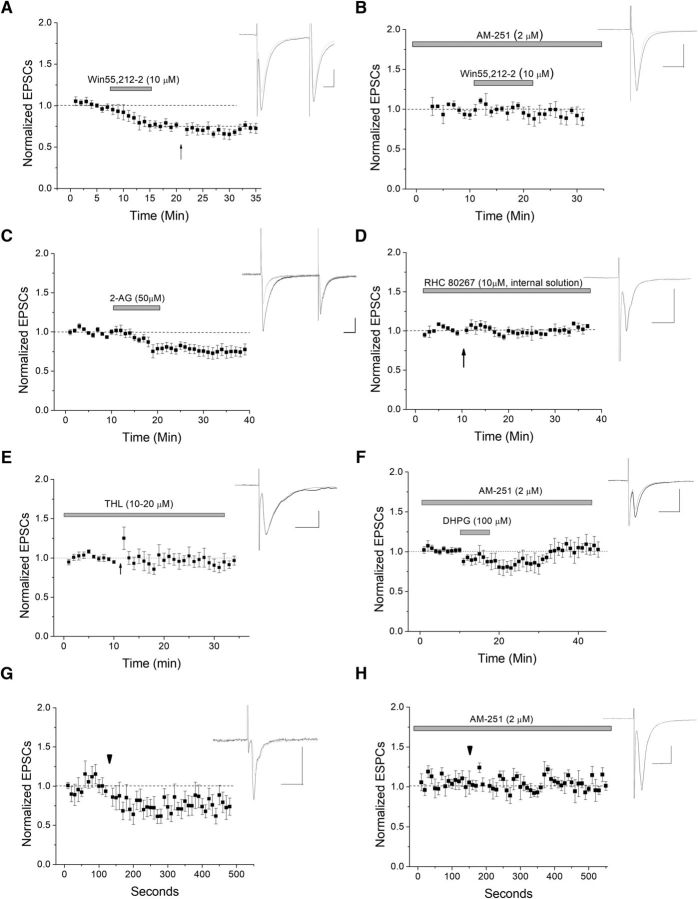
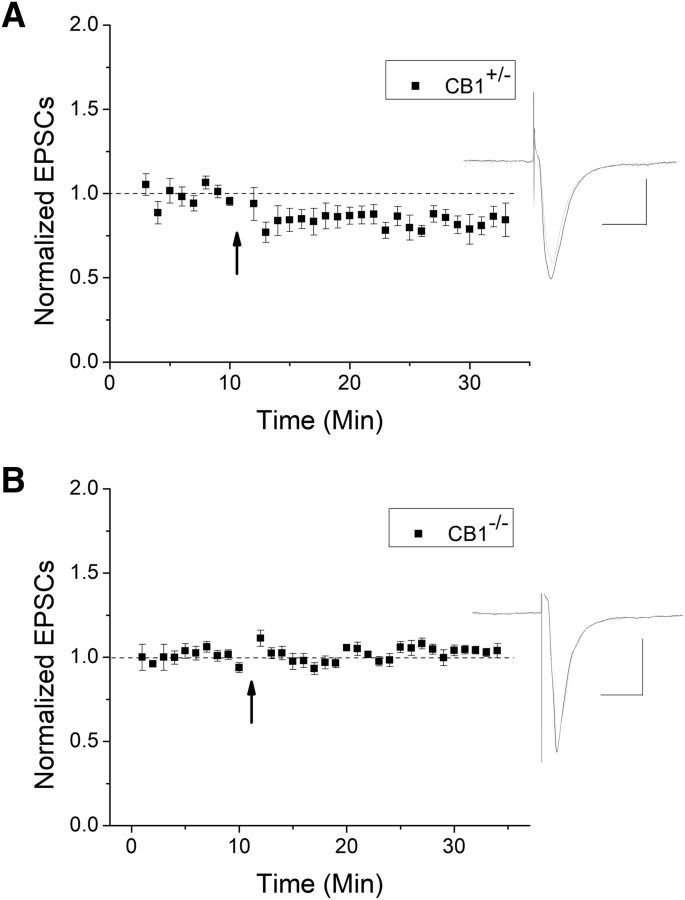
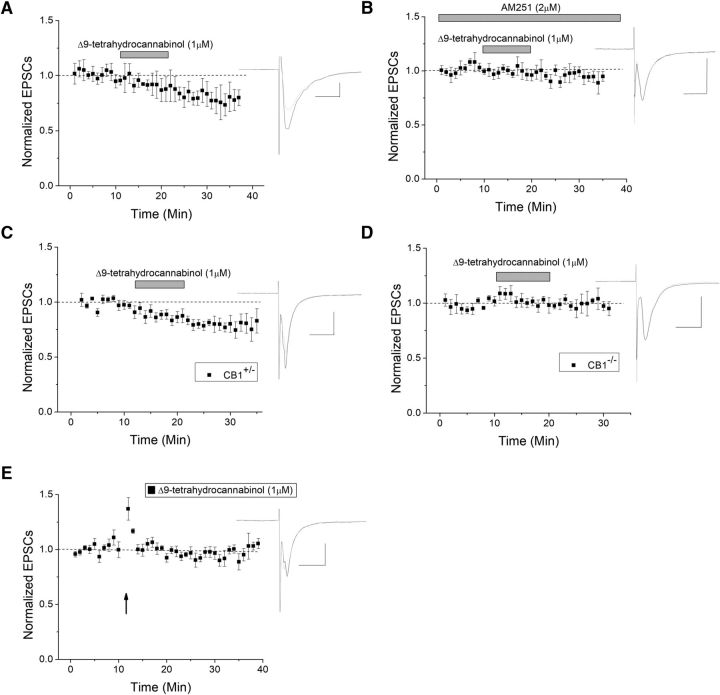
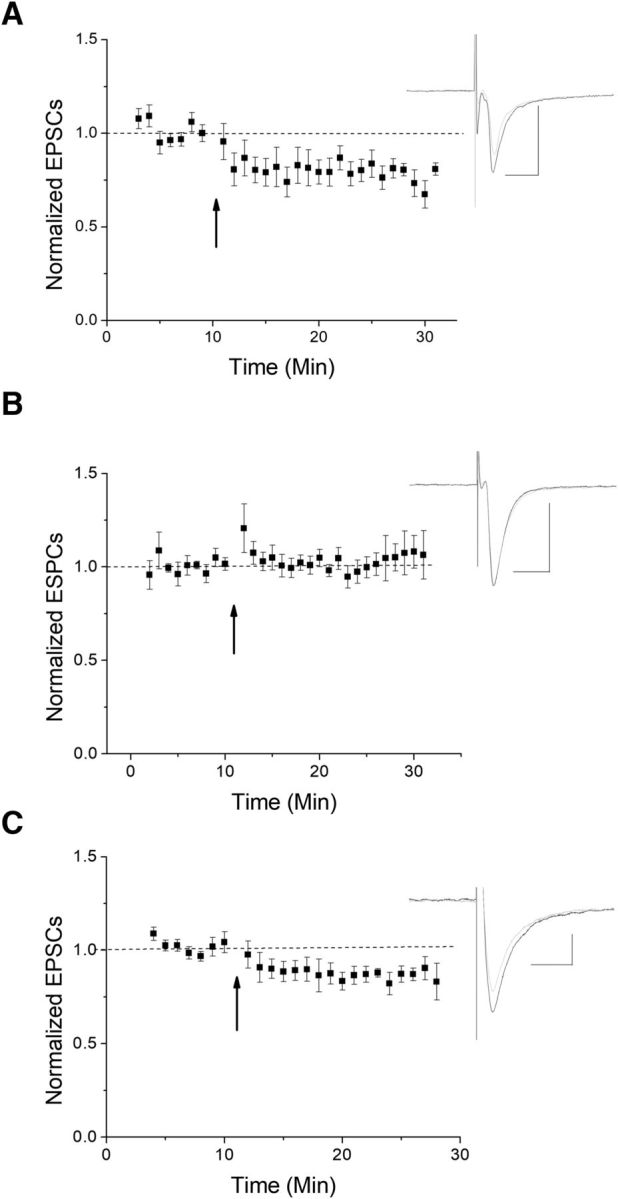
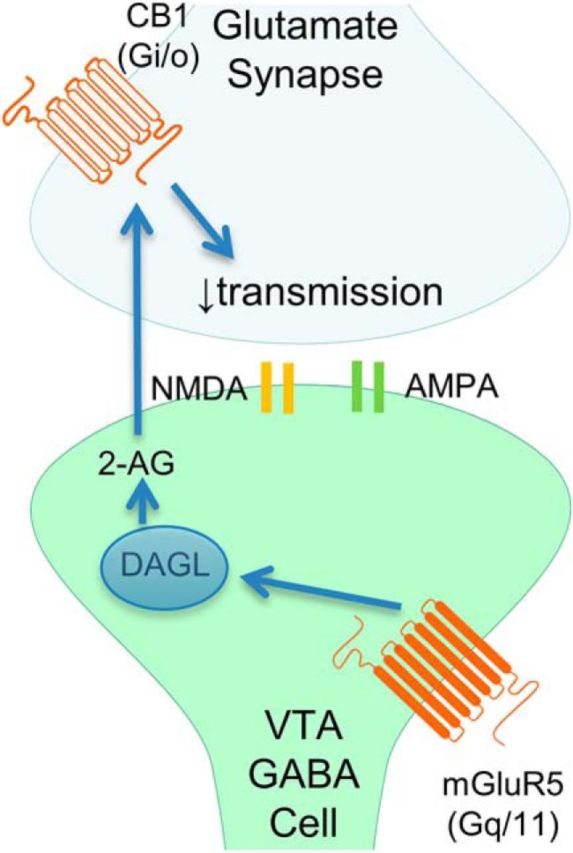
The VTA is necessary for reward behavior with dopamine cells critically involved in reward signaling. Dopamine cells in turn are innervated and regulated by neighboring inhibitory GABA cells. Using whole-cell electrophysiology in juvenile-adolescent GAD67-GFP male mice, we examined excitatory plasticity in fluorescent VTA GABA cells. A novel CB1-dependent LTD was induced in GABA cells that was dependent on metabotropic glutamate receptor 5, and cannabinoid receptor 1 (CB1). LTD was absent in CB1 knock-out mice but preserved in heterozygous littermates. Bath applied Δ9-tetrahydrocannabinol depressed GABA cell activity, therefore downstream dopamine cells will be disinhibited; and thus, this could potentially result in increased reward. Chronic injections of Δ9-tetrahydrocannabinol occluded LTD compared with vehicle injections; however, a single exposure was insufficient to do so. As synaptic modifications by drugs of abuse are often tied to addiction, these data suggest a possible mechanism for the addictive effects of Δ9-tetrahydrocannabinol in juvenile-adolescents, by potentially altering reward behavioral outcomes.SIGNIFICANCE STATEMENT The present study identifies a novel form of glutamatergic synaptic plasticity in VTA GABA neurons, a currently understudied cell type that is critical for the brain's reward circuit, and how Δ9-tetrahydrocannabinol occludes this plasticity. This study specifically addresses a potential unifying mechanism whereby marijuana could exert rewarding and addictive/withdrawal effects. Marijuana use and legalization are a pressing issue for many states in the United States. Although marijuana is the most commonly abused illicit drug, the implications of legalized, widespread, or continued usage are speculative. This study in juvenile-adolescent aged mice identifies a novel form of synaptic plasticity in VTA GABA cells, and the synaptic remodeling that can occur after Δ9-tetrahydrocannabinol use.
Keywords: THC; TRPV1; anandamide; endocannabinoid; mGluR5; withdrawal.
Copyright © 2017 the authors 0270-6474/17/3710943-12$15.00/0.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials