

Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases
- PMID: 29038287
- PMCID: PMC5748917
- DOI: 10.1681/ASN.2017050483
Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases
Abstract
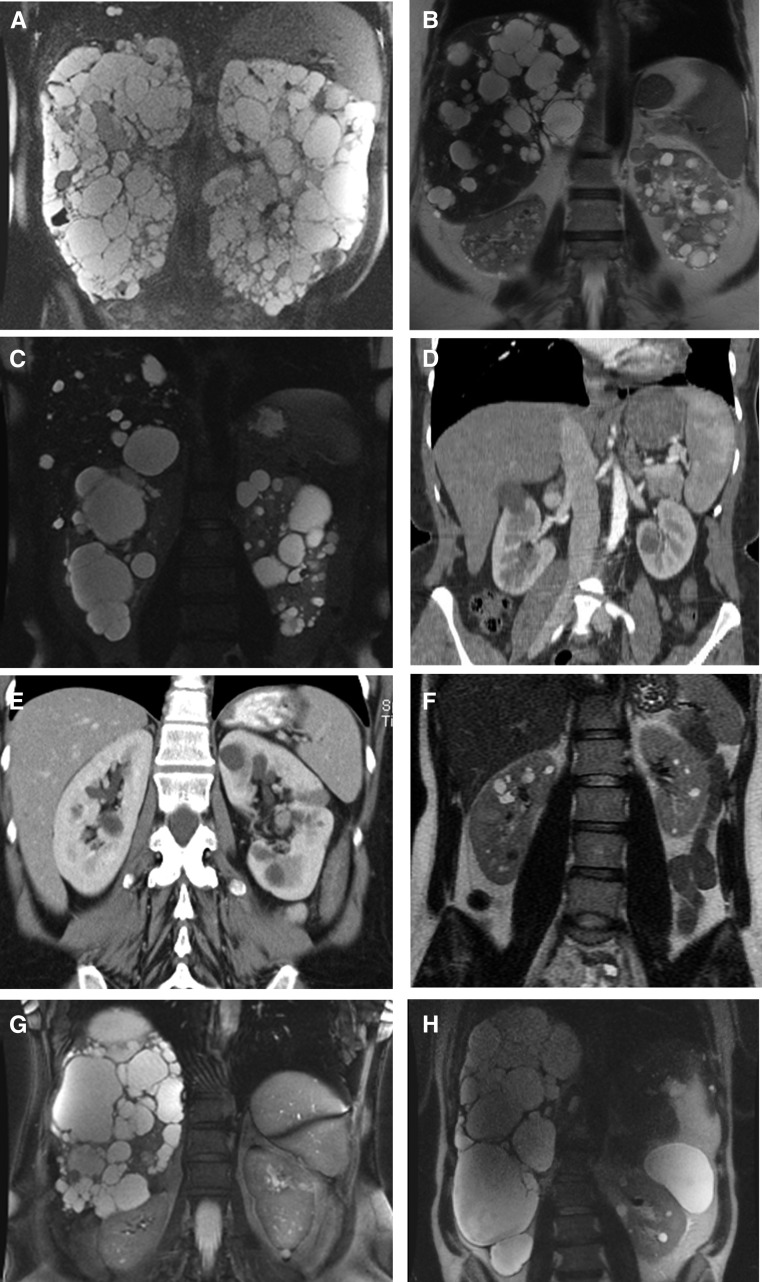
Data indicate significant phenotypic and genotypic overlap, plus a common pathogenesis, between two groups of inherited disorders, autosomal dominant polycystic kidney diseases (ADPKD), a significant cause of ESRD, and autosomal dominant polycystic liver diseases (ADPLD), which result in significant PLD with minimal PKD. Eight genes have been associated with ADPKD (PKD1 and PKD2), ADPLD (PRKCSH, SEC63, LRP5, ALG8, and SEC61B), or both (GANAB). Although genetics is only infrequently used for diagnosing these diseases and prognosing the associated outcomes, its value is beginning to be appreciated, and the genomics revolution promises more reliable and less expensive molecular diagnostic tools for these diseases. We therefore propose categorization of patients with a phenotypic and genotypic descriptor that will clarify etiology, provide prognostic information, and better describe atypical cases. In genetically defined cases, the designation would include the disease and gene names, with allelic (truncating/nontruncating) information included for PKD1 Recent data have shown that biallelic disease including at least one weak ADPKD allele is a significant cause of symptomatic, very early onset ADPKD. Including a genic (and allelic) descriptor with the disease name will provide outcome clues, guide treatment, and aid prevalence estimates.
Keywords: ADPKD; cystic kidney; liver cysts; polycystic kidney disease.
Copyright © 2018 by the American Society of Nephrology.
Figures
References
-
- Ong AC, Devuyst O, Knebelmann B, Walz G; ERA-EDTA Working Group for Inherited Kidney Diseases : Autosomal dominant polycystic kidney disease: The changing face of clinical management. Lancet 385: 1993–2002, 2015 - PubMed
-
- Gabow PA, Johnson AM, Kaehny WD, Kimberling WJ, Lezotte DC, Duley IT, Jones RH: Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 41: 1311–1319, 1992 - PubMed
-
- Audrézet MP, Corbiere C, Lebbah S, Morinière V, Broux F, Louillet F, Fischbach M, Zaloszyc A, Cloarec S, Merieau E, Baudouin V, Deschênes G, Roussey G, Maestri S, Visconti C, Boyer O, Abel C, Lahoche A, Randrianaivo H, Bessenay L, Mekahli D, Ouertani I, Decramer S, Ryckenwaert A, Cornec-Le Gall E, Salomon R, Ferec C, Heidet L: Comprehensive PKD1 and PKD2 mutation analysis in prenatal autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 722–729, 2016 - PMC - PubMed
-
- Fick GM, Johnson AM, Strain JD, Kimberling WJ, Kumar S, Manco-Johnson ML, Duley IT, Gabow PA: Characteristics of very early onset autosomal dominant polycystic kidney disease. J Am Soc Nephrol 3: 1863–1870, 1993 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
