

Mutation-specific downregulation of CFTR2 variants by gating potentiators
- PMID: 29040544
- PMCID: PMC5886047
- DOI: 10.1093/hmg/ddx367
Mutation-specific downregulation of CFTR2 variants by gating potentiators
Abstract
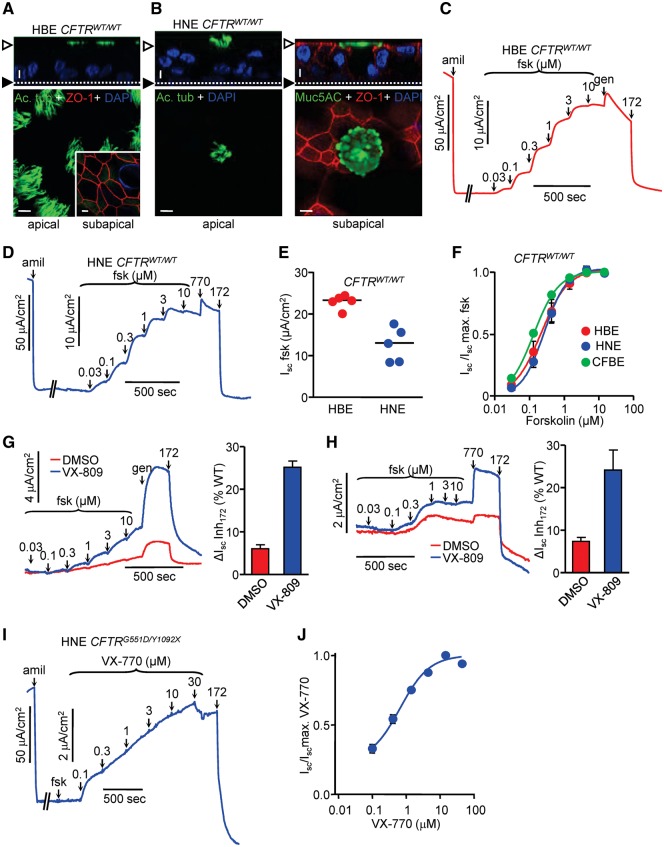
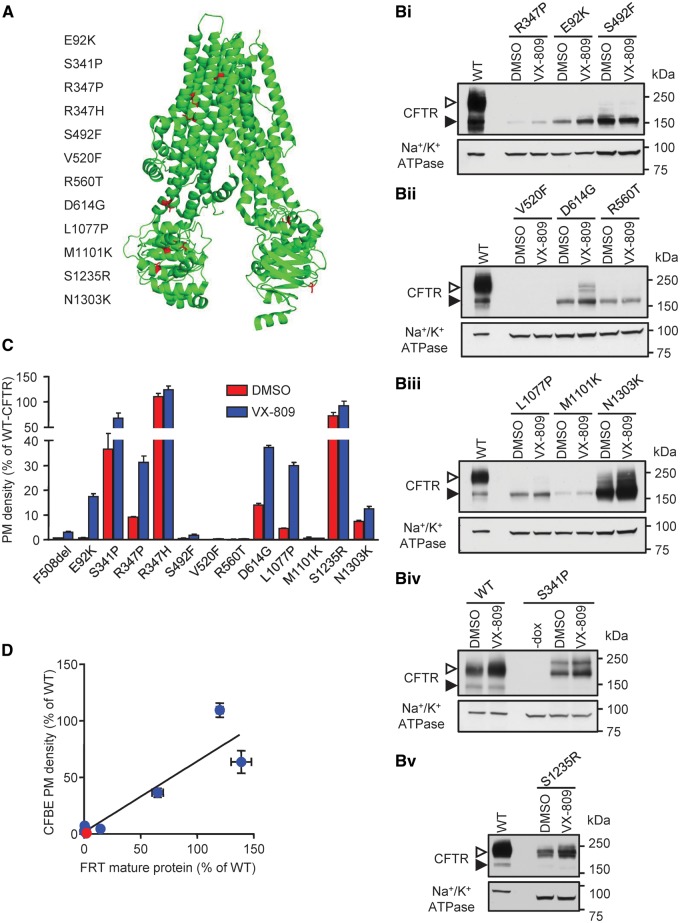
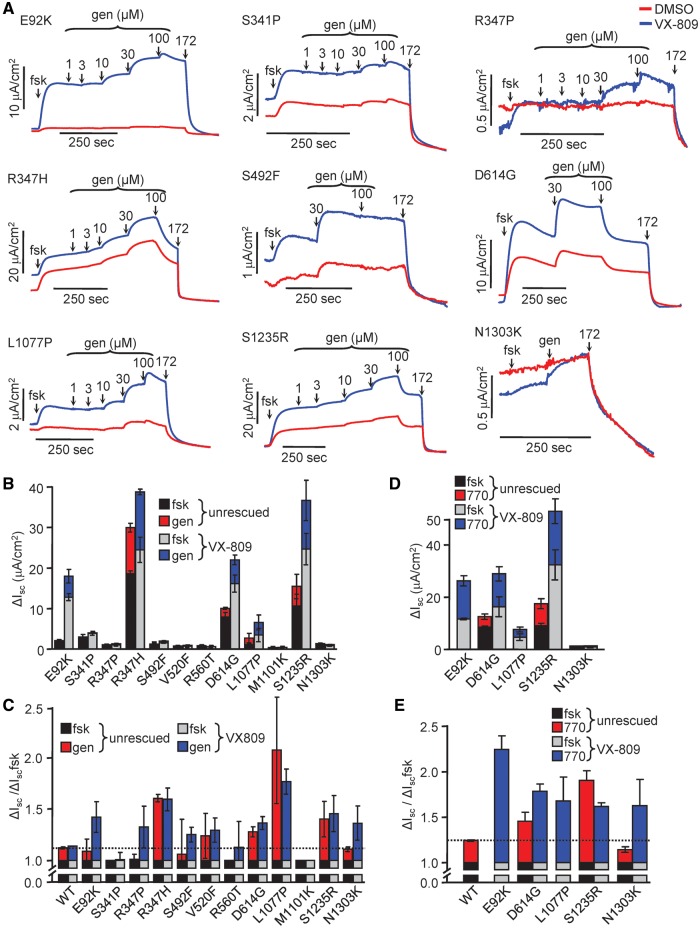
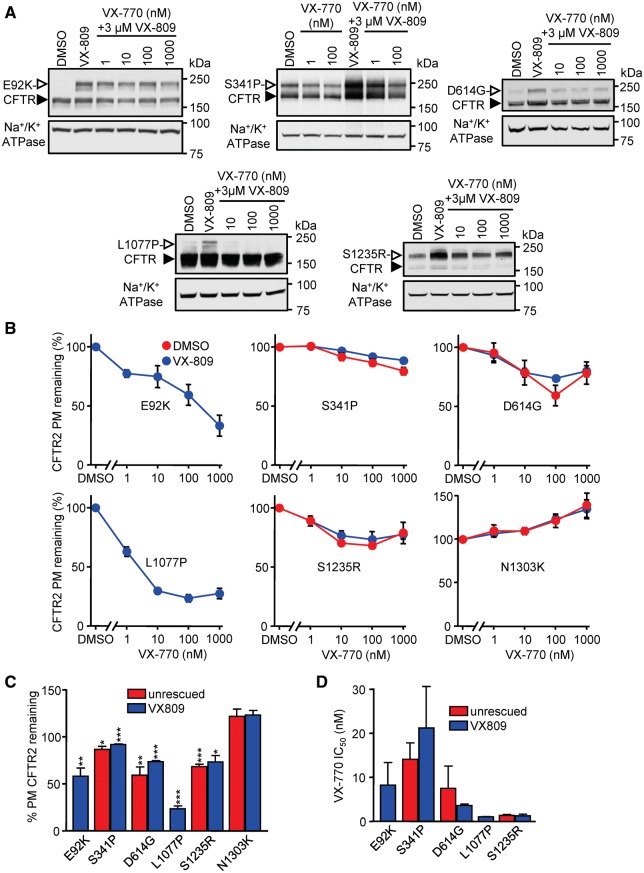
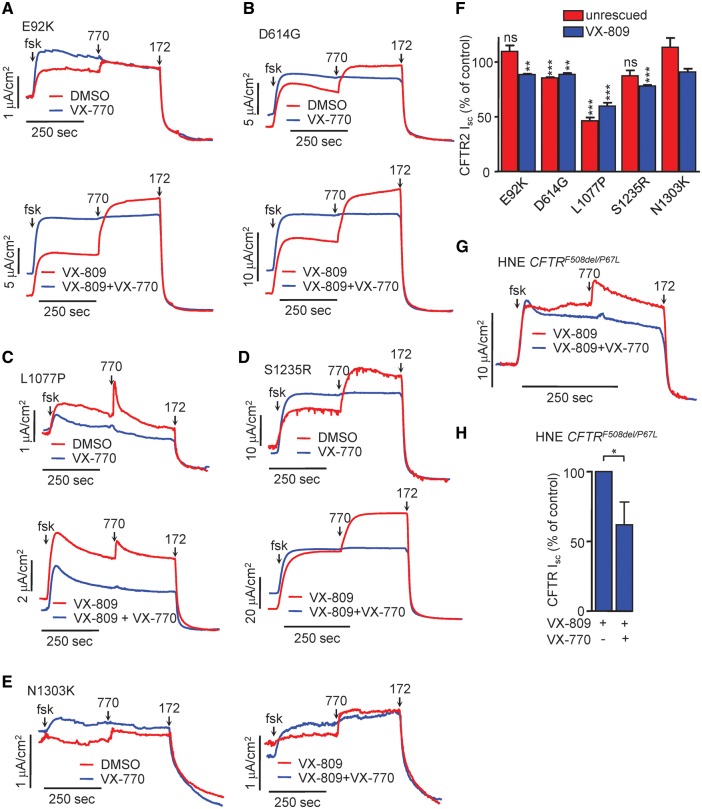
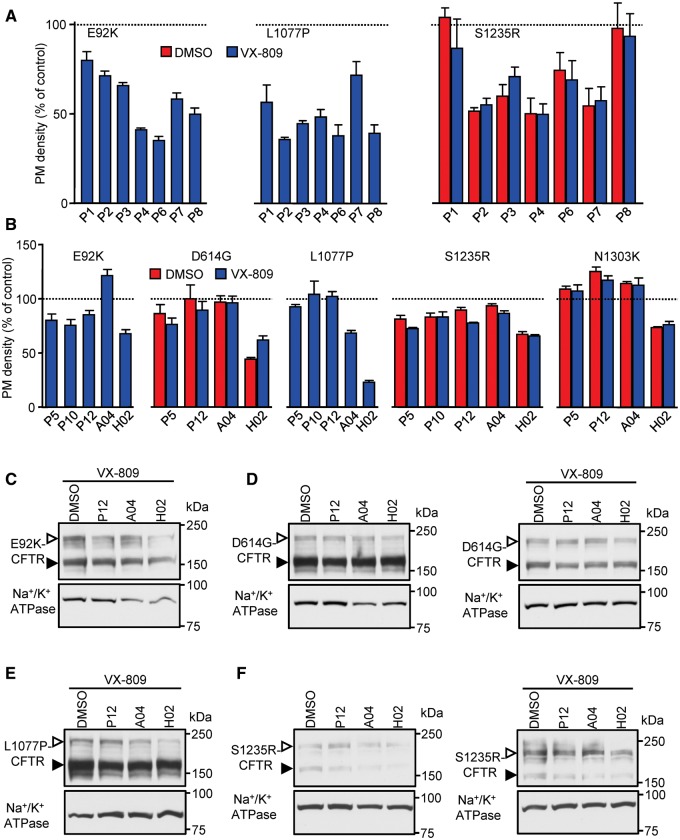
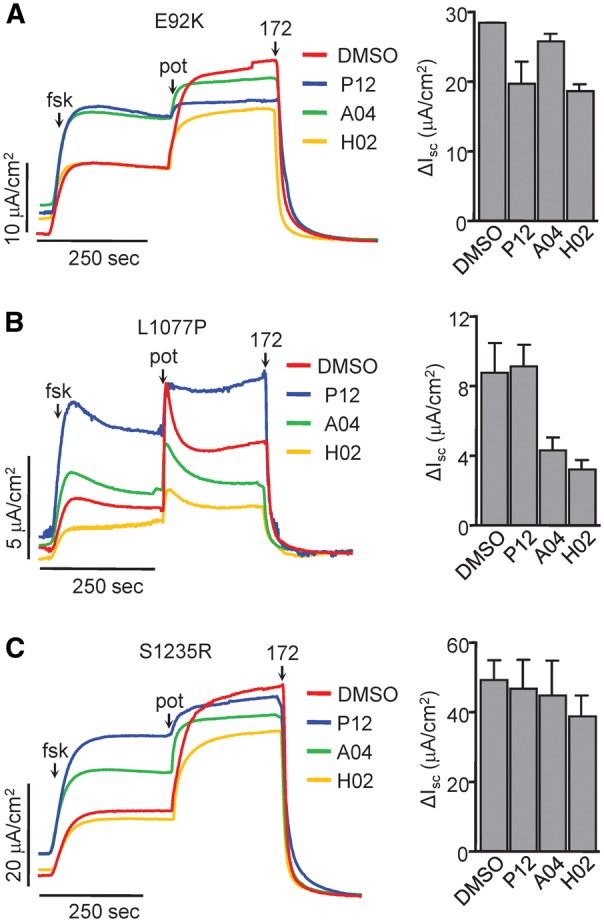
Approximately 50% of cystic fibrosis (CF) patients are heterozygous with a rare mutation on at least one allele. Several mutants exhibit functional defects, correctable by gating potentiators. Long-term exposure (≥24 h) to the only available potentiator drug, VX-770, leads to the biochemical and functional downregulation of F508del-CFTR both in immortalized and primary human airway cells, and possibly other CF mutants, attenuating its beneficial effect. Based on these considerations, we wanted to determine the effect of chronic VX-770 exposure on the functional and biochemical expression of rare CF processing/gating mutants in human airway epithelia. Expression of CFTR2 mutants was monitored in the human bronchial epithelial cell line (CFBE41o-) and in patient-derived conditionally reprogrammed bronchial and nasal epithelia by short-circuit current measurements, cell surface ELISA and immunoblotting in the absence or presence of CFTR modulators. The VX-770 half-maximal effective (EC50) concentration for G551D-CFTR activation was ∼0.63 μM in human nasal epithelia, implying that comparable concentration is required in the lung to attain clinical benefit. Five of the twelve rare CFTR2 mutants were susceptible to ∼20-70% downregulation by chronic VX-770 exposure with an IC50 of ∼1-20 nM and to destabilization by other investigational potentiators, thereby diminishing the primary functional gain of CFTR modulators. Thus, chronic exposure to VX-770 and preclinical potentiators can destabilize CFTR2 mutants in human airway epithelial models in a mutation and compound specific manner. This highlights the importance of selecting potentiator drugs with minimal destabilizing effects on CF mutants, advocating a precision medicine approach.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Rowe S.M., Miller S., Sorscher E.J. (2005) Cystic fibrosis. N. Engl. J. Med., 352, 1992–2001. - PubMed
-
- Riordan J.R. (2008) CFTR function and prospects for therapy. Annu. Rev. Biochem., 77, 701–726. - PubMed
-
- CFTR2 Database. Available: http://www.cftr2.org; date last accessed September 27, 2017.
-
- Cystic Fibrosis Mutation Database. Available: http://www.genet.sickkids.on.ca/app; date last accessed September 27, 2017.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
