

Towards a balanced view of the bacterial tree of life
- PMID: 29041958
- PMCID: PMC5644168
- DOI: 10.1186/s40168-017-0360-9
Towards a balanced view of the bacterial tree of life
Erratum in
-
Correction to: Towards a balanced view of the bacterial tree of life.Microbiome. 2017 Nov 15;5(1):149. doi: 10.1186/s40168-017-0367-2. Microbiome. 2017. PMID: 29141685 Free PMC article.
Abstract
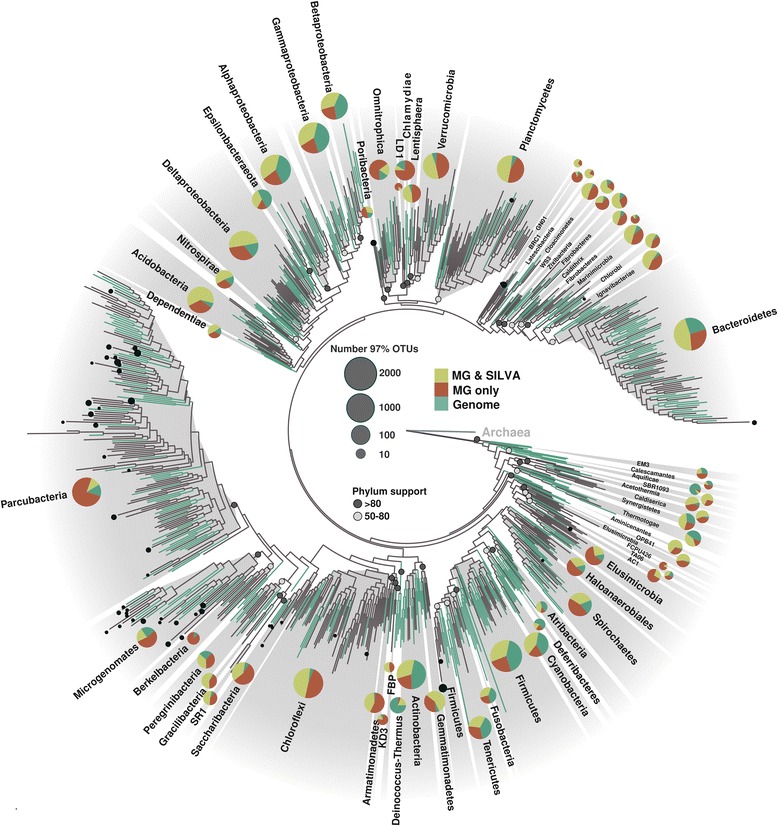
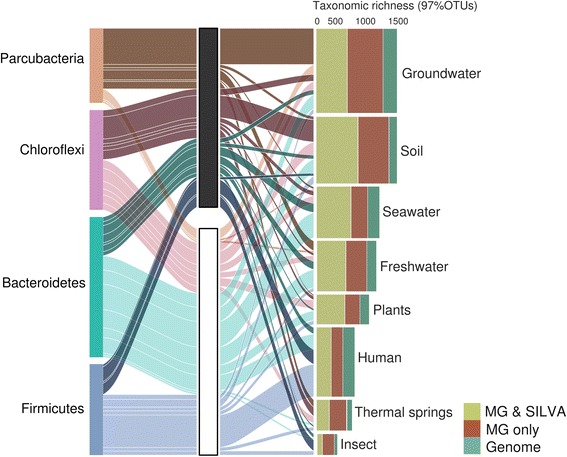
The bacterial tree of life has recently undergone significant expansion, chiefly from candidate phyla retrieved through genome-resolved metagenomics. Bypassing the need for genome availability, we present a snapshot of bacterial phylogenetic diversity based on the recovery of high-quality SSU rRNA gene sequences extracted from nearly 7000 metagenomes and all available reference genomes. We illuminate taxonomic richness within established bacterial phyla together with environmental distribution patterns, providing a revised framework for future phylogeny-driven sequencing efforts.
Keywords: Bacteria; Bacterial diversity; Candidate Phyla Radiation (CPR); Metagenomics; Microbial dark matter (MDM); Novel bacterial lineages; Small subunit (SSU) rRNA; Tree of life.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing financial or non-financial interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Metagenomics uncovers gaps in amplicon-based detection of microbial diversity.Nat Microbiol. 2016 Feb 1;1:15032. doi: 10.1038/nmicrobiol.2015.32. Nat Microbiol. 2016. PMID: 27572438
-
Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life.Nat Microbiol. 2017 Nov;2(11):1533-1542. doi: 10.1038/s41564-017-0012-7. Epub 2017 Sep 11. Nat Microbiol. 2017. PMID: 28894102
-
Major New Microbial Groups Expand Diversity and Alter our Understanding of the Tree of Life.Cell. 2018 Mar 8;172(6):1181-1197. doi: 10.1016/j.cell.2018.02.016. Cell. 2018. PMID: 29522741 Review.
-
Members of the Candidate Phyla Radiation are functionally differentiated by carbon- and nitrogen-cycling capabilities.Microbiome. 2017 Sep 2;5(1):112. doi: 10.1186/s40168-017-0331-1. Microbiome. 2017. PMID: 28865481 Free PMC article.
-
High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota.Gut. 2008 Nov;57(11):1605-15. doi: 10.1136/gut.2007.133603. Gut. 2008. PMID: 18941009 Review.
Cited by
-
Exploring Andean High-Altitude Lake Extremophiles through Advanced Proteotyping.J Proteome Res. 2024 Mar 1;23(3):891-904. doi: 10.1021/acs.jproteome.3c00538. Epub 2024 Feb 20. J Proteome Res. 2024. PMID: 38377575 Free PMC article.
-
A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life.Nat Biotechnol. 2018 Nov;36(10):996-1004. doi: 10.1038/nbt.4229. Epub 2018 Aug 27. Nat Biotechnol. 2018. PMID: 30148503
-
A Systematic Approach to Bacterial Phylogeny Using Order Level Sampling and Identification of HGT Using Network Science.Microorganisms. 2020 Feb 24;8(2):312. doi: 10.3390/microorganisms8020312. Microorganisms. 2020. PMID: 32102454 Free PMC article.
-
Identification and characterization of a novel chromosomal aminoglycoside 3'-O-phosphotransferase, APH(3')-Id, from Kluyvera intermedia DW18 isolated from the sewage of an animal farm.Front Microbiol. 2023 Aug 28;14:1224464. doi: 10.3389/fmicb.2023.1224464. eCollection 2023. Front Microbiol. 2023. PMID: 37700861 Free PMC article.
-
Candidate Phyla Radiation Roizmanbacteria From Hot Springs Have Novel and Unexpectedly Abundant CRISPR-Cas Systems.Front Microbiol. 2019 May 3;10:928. doi: 10.3389/fmicb.2019.00928. eCollection 2019. Front Microbiol. 2019. PMID: 31130929 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
