

RECQ-like helicases Sgs1 and BLM regulate R-loop-associated genome instability
- PMID: 29042409
- PMCID: PMC5716281
- DOI: 10.1083/jcb.201703168
RECQ-like helicases Sgs1 and BLM regulate R-loop-associated genome instability
Abstract
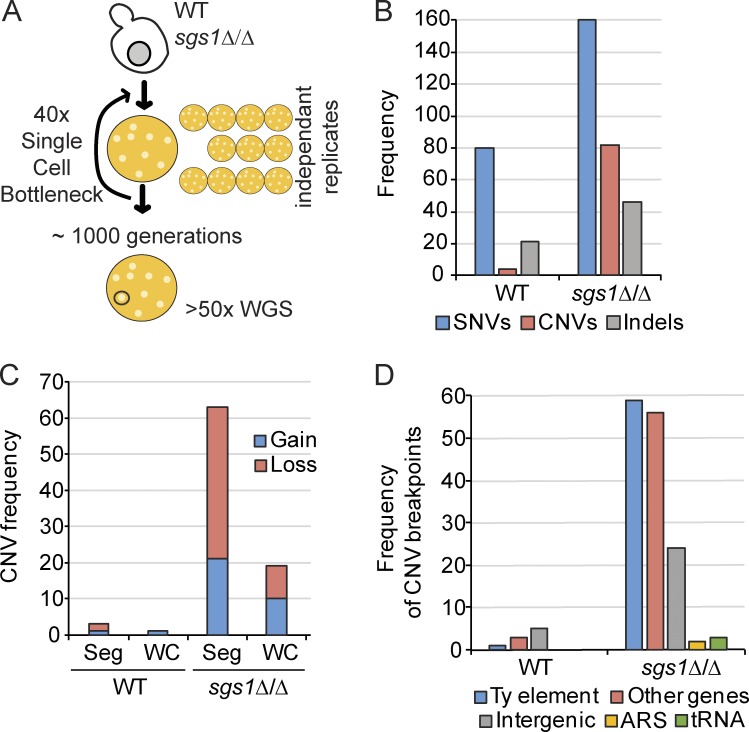
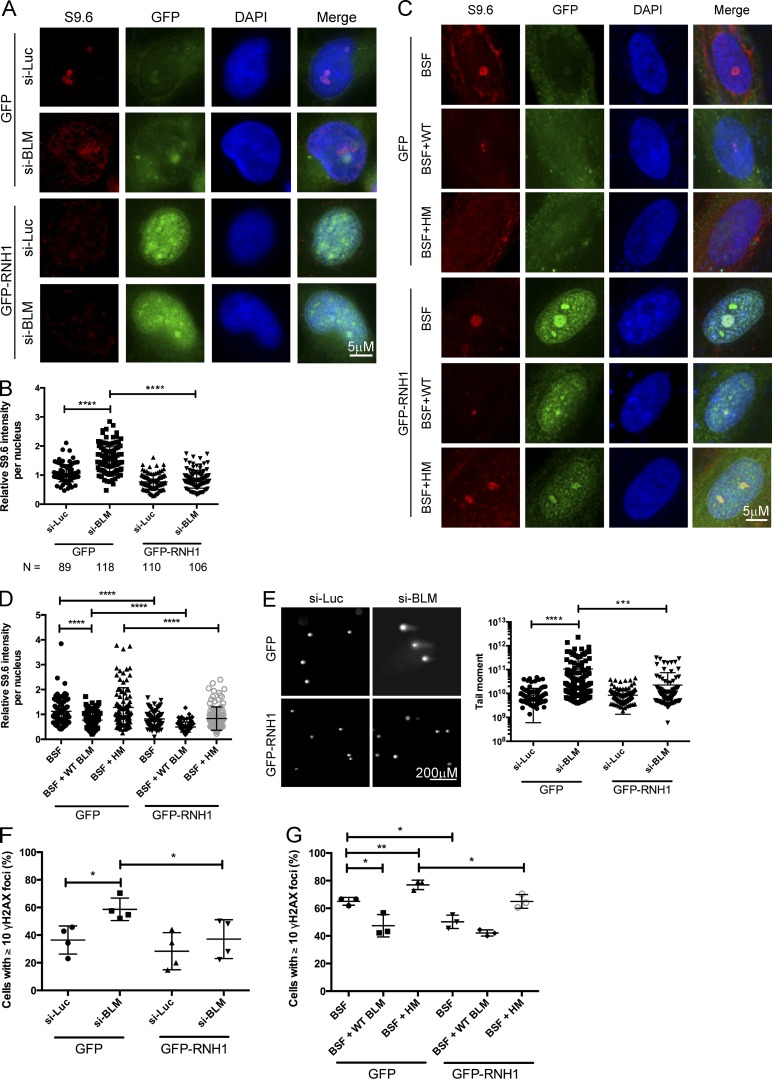
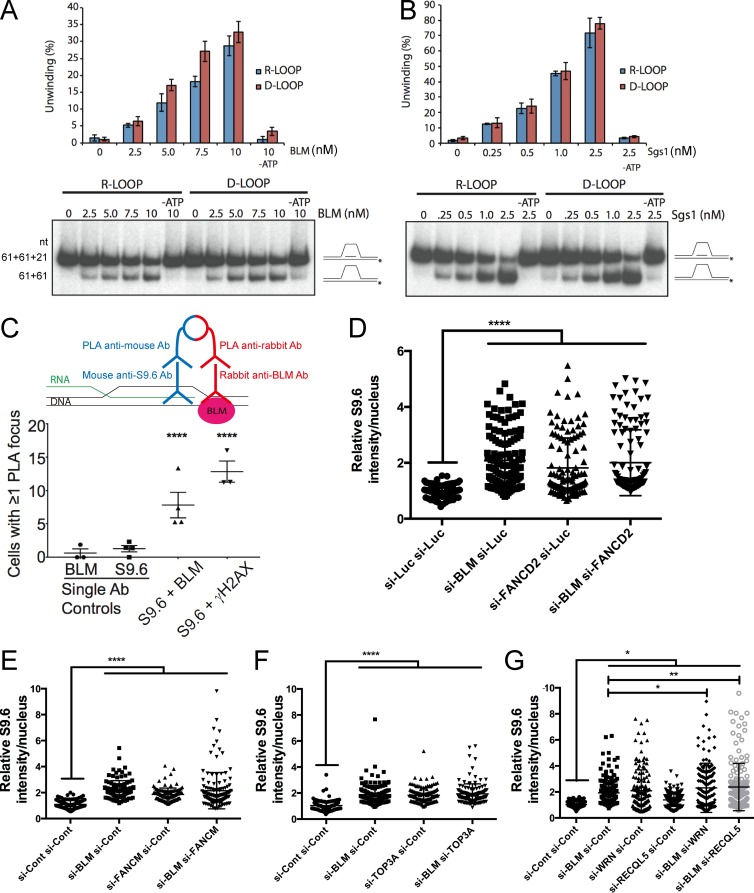
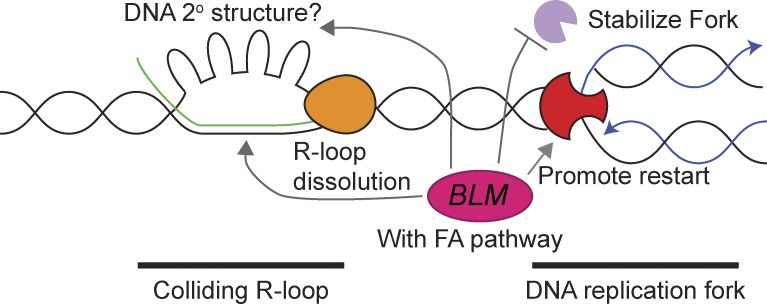
Sgs1, the orthologue of human Bloom's syndrome helicase BLM, is a yeast DNA helicase functioning in DNA replication and repair. We show that SGS1 loss increases R-loop accumulation and sensitizes cells to transcription-replication collisions. Yeast lacking SGS1 accumulate R-loops and γ-H2A at sites of Sgs1 binding, replication pausing regions, and long genes. The mutation signature of sgs1Δ reveals copy number changes flanked by repetitive regions with high R-loop-forming potential. Analysis of BLM in Bloom's syndrome fibroblasts or by depletion of BLM from human cancer cells confirms a role for Sgs1/BLM in suppressing R-loop-associated genome instability across species. In support of a potential direct effect, BLM is found physically proximal to DNA:RNA hybrids in human cells, and can efficiently unwind R-loops in vitro. Together, our data describe a conserved role for Sgs1/BLM in R-loop suppression and support an increasingly broad view of DNA repair and replication fork stabilizing proteins as modulators of R-loop-mediated genome instability.
© 2017 Chang et al.
Figures
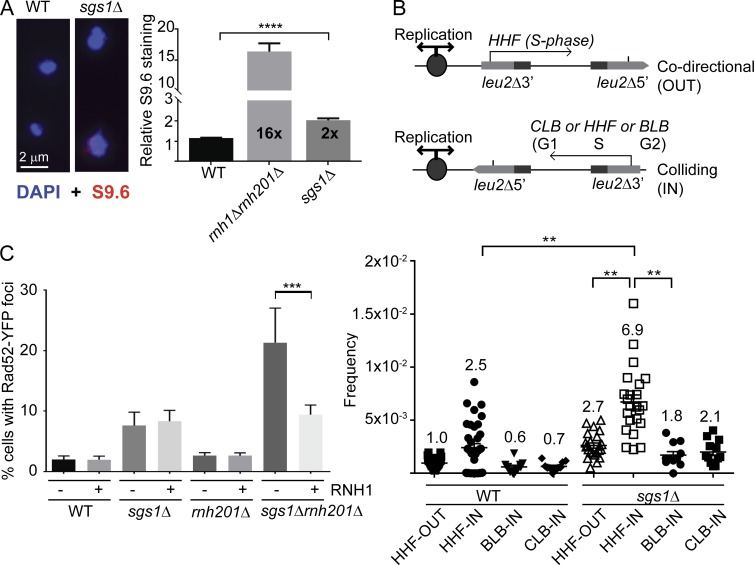
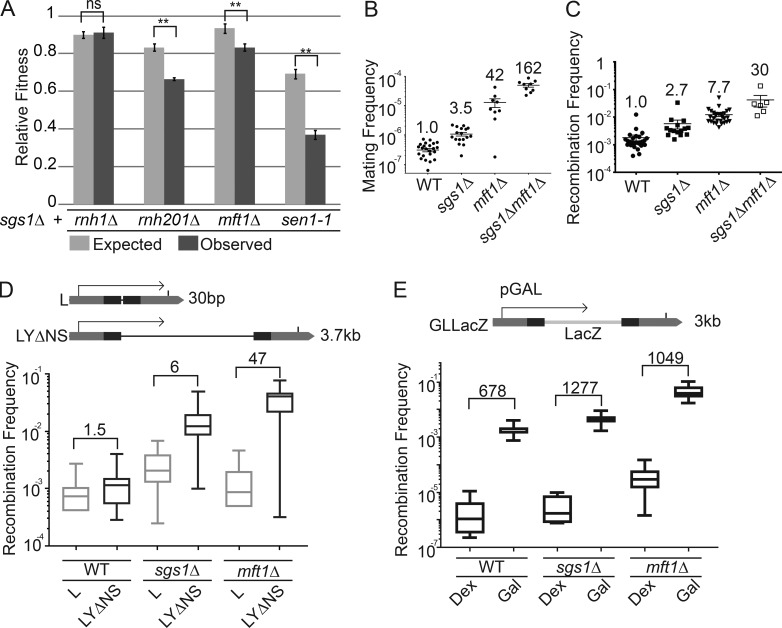
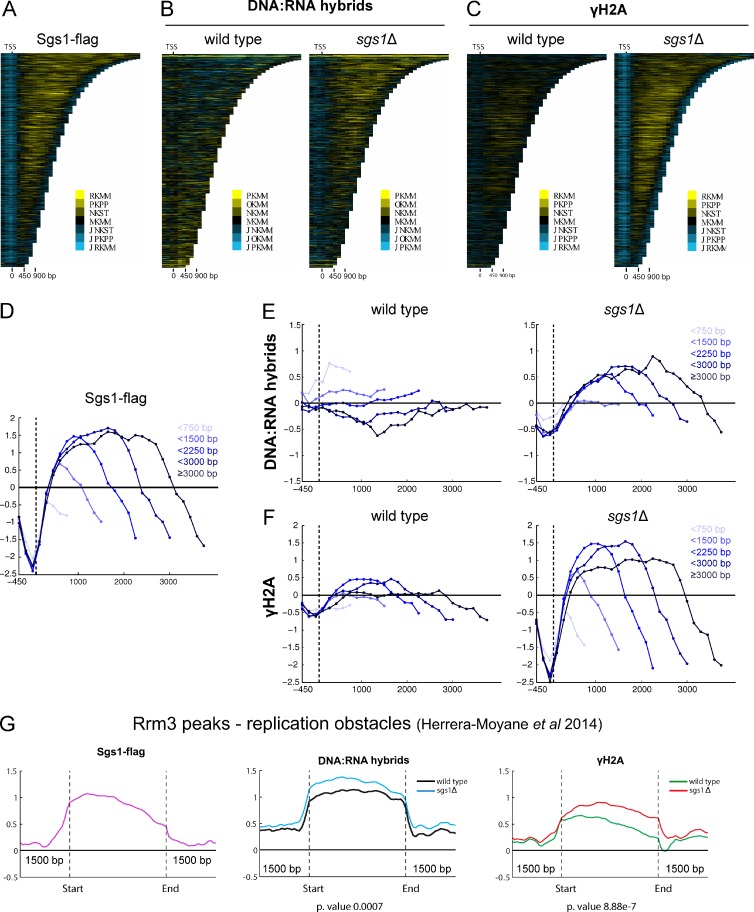
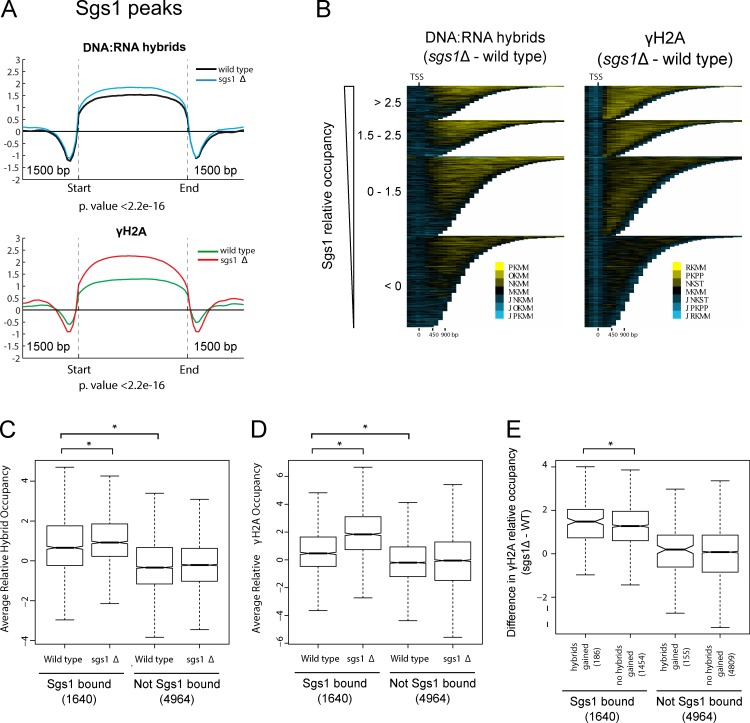
Similar articles
-
Sgs1 truncations induce genome rearrangements but suppress detrimental effects of BLM overexpression in Saccharomyces cerevisiae.J Mol Biol. 2011 Jan 28;405(4):877-91. doi: 10.1016/j.jmb.2010.11.035. Epub 2010 Nov 25. J Mol Biol. 2011. PMID: 21111748 Free PMC article.
-
Bloom's and Werner's syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases.Proc Natl Acad Sci U S A. 1998 Jul 21;95(15):8733-8. doi: 10.1073/pnas.95.15.8733. Proc Natl Acad Sci U S A. 1998. PMID: 9671747 Free PMC article.
-
The RecQ helicase Sgs1 drives ATP-dependent disruption of Rad51 filaments.Nucleic Acids Res. 2019 May 21;47(9):4694-4706. doi: 10.1093/nar/gkz186. Nucleic Acids Res. 2019. PMID: 30916344 Free PMC article.
-
[Functional analysis of yeast homologue gene associated with human DNA helicase causative syndromes].Kokuritsu Iyakuhin Shokuhin Eisei Kenkyusho Hokoku. 2002;(120):53-74. Kokuritsu Iyakuhin Shokuhin Eisei Kenkyusho Hokoku. 2002. PMID: 12638184 Review. Japanese.
-
Maintenance of Yeast Genome Integrity by RecQ Family DNA Helicases.Genes (Basel). 2020 Feb 18;11(2):205. doi: 10.3390/genes11020205. Genes (Basel). 2020. PMID: 32085395 Free PMC article. Review.
Cited by
-
RNA biogenesis and RNA metabolism factors as R-loop suppressors: a hidden role in genome integrity.Genes Dev. 2024 Jul 19;38(11-12):504-527. doi: 10.1101/gad.351853.124. Genes Dev. 2024. PMID: 38986581 Free PMC article. Review.
-
Walking a tightrope: The complex balancing act of R-loops in genome stability.Mol Cell. 2022 Jun 16;82(12):2267-2297. doi: 10.1016/j.molcel.2022.04.014. Epub 2022 May 3. Mol Cell. 2022. PMID: 35508167 Free PMC article. Review.
-
ATM pathway activation limits R-loop-associated genomic instability in Werner syndrome cells.Nucleic Acids Res. 2019 Apr 23;47(7):3485-3502. doi: 10.1093/nar/gkz025. Nucleic Acids Res. 2019. PMID: 30657978 Free PMC article.
-
Transcription/Replication Conflicts in Tumorigenesis and Their Potential Role as Novel Therapeutic Targets in Multiple Myeloma.Cancers (Basel). 2021 Jul 27;13(15):3755. doi: 10.3390/cancers13153755. Cancers (Basel). 2021. PMID: 34359660 Free PMC article. Review.
-
BLM mutation is associated with increased tumor mutation burden and improved survival after immunotherapy across multiple cancers.Cancer Med. 2024 Jan;13(1):e6716. doi: 10.1002/cam4.6716. Epub 2023 Dec 20. Cancer Med. 2024. PMID: 38124443 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
