

Thrombotic Microangiopathy and the Kidney
- PMID: 29042465
- PMCID: PMC5967417
- DOI: 10.2215/CJN.00620117
Thrombotic Microangiopathy and the Kidney
Abstract
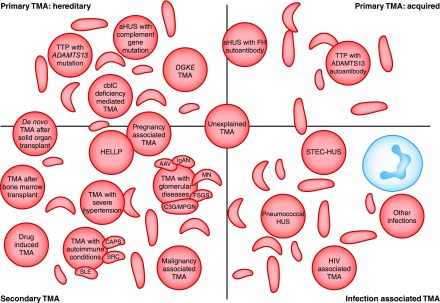
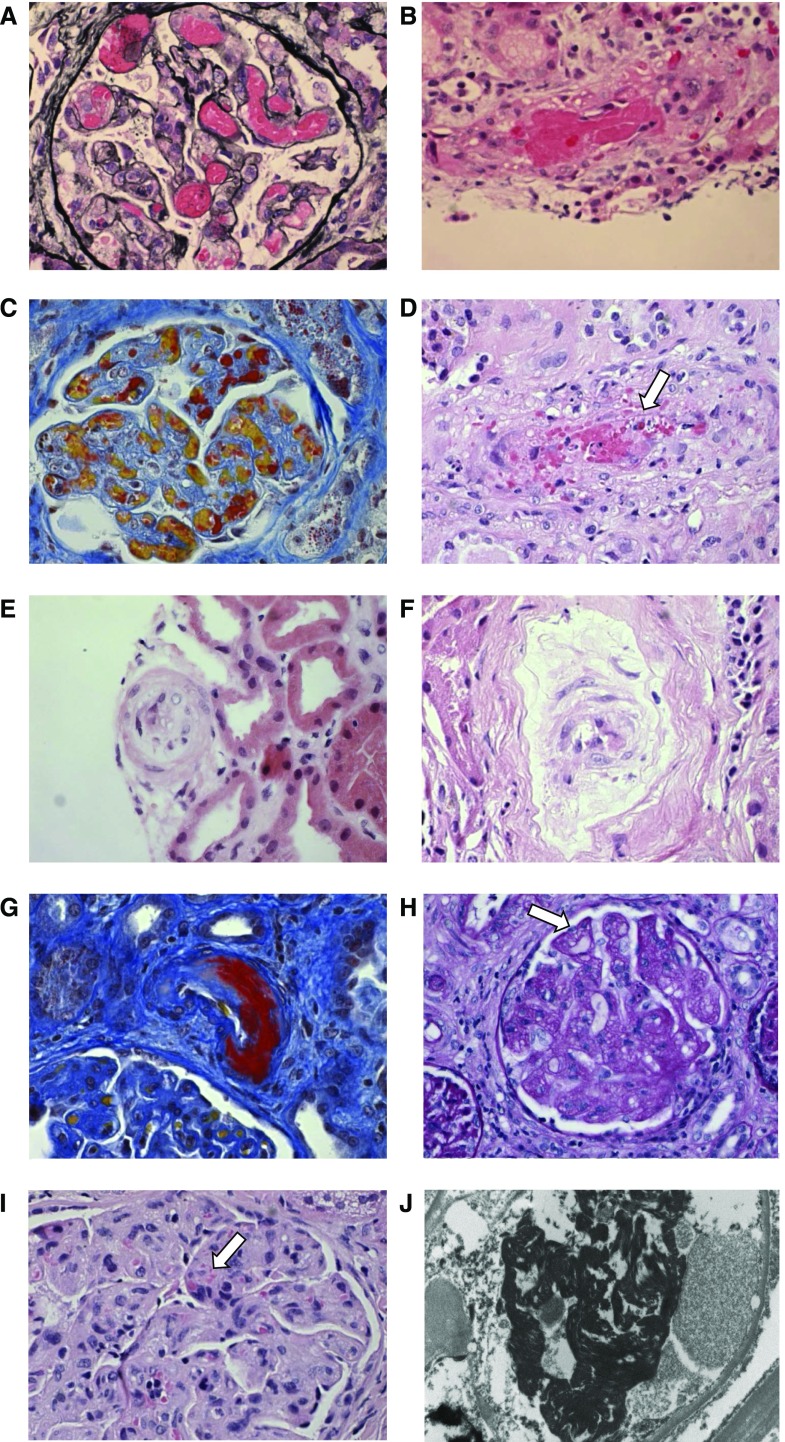
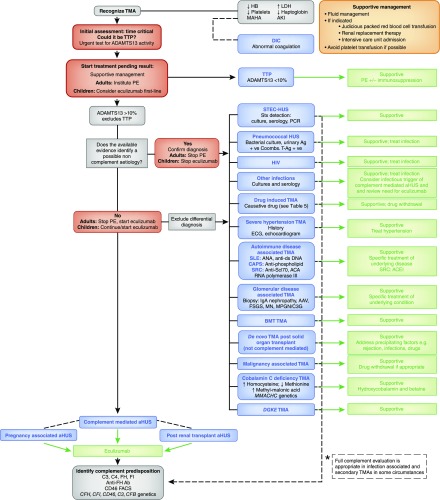
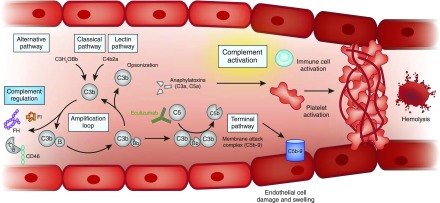
Thrombotic microangiopathy can manifest in a diverse range of diseases and is characterized by thrombocytopenia, microangiopathic hemolytic anemia, and organ injury, including AKI. It can be associated with significant morbidity and mortality, but a systematic approach to investigation and prompt initiation of supportive management and, in some cases, effective specific treatment can result in good outcomes. This review considers the classification, pathology, epidemiology, characteristics, and pathogenesis of the thrombotic microangiopathies, and outlines a pragmatic approach to diagnosis and management.
Keywords: Acute Kidney Injury; Anemia; Complement; Hemolytic; Purpura; Thrombotic Microangiopathies; Thrombotic Thrombocytopenic; atypical hemolytic uremic syndrome; glomerular disease; kidney.
Copyright © 2018 by the American Society of Nephrology.
Figures
References
-
- Moake JL: Thrombotic microangiopathies. N Engl J Med 347: 589–600, 2002 - PubMed
-
- George JN, Nester CM: Syndromes of thrombotic microangiopathy. N Engl J Med 371: 654–666, 2014 - PubMed
-
- Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, Kavanagh D, Noris M, Pickering M, Sanchez-Corral P, Skerka C, Zipfel P, Smith RJ: Atypical aHUS: State of the art. Mol Immunol 67: 31–42, 2015 - PubMed
-
- Scully M, Cataland S, Coppo P, de la Rubia J, Friedman KD, Kremer Hovinga J, Lammle B, Matsumoto M, Pavenski K, Sadler E, Sarode R, Wu H; International Working Group for Thrombotic Thrombocytopenic Purpura: Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost 15: 312–322, 2017. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
