

Insights from the Genome Sequence of Mycobacterium lepraemurium: Massive Gene Decay and Reductive Evolution
- PMID: 29042494
- PMCID: PMC5646247
- DOI: 10.1128/mBio.01283-17
Insights from the Genome Sequence of Mycobacterium lepraemurium: Massive Gene Decay and Reductive Evolution
Abstract
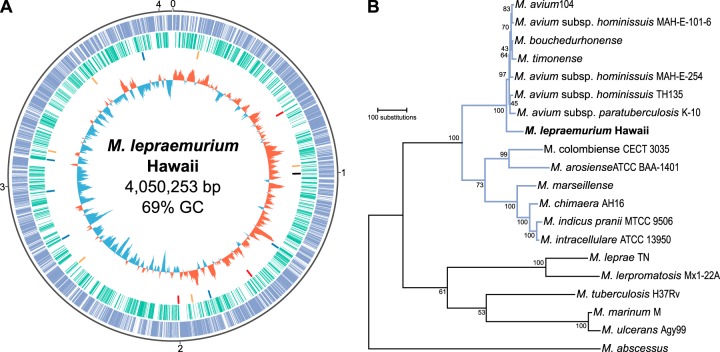
Mycobacterium lepraemurium is the causative agent of murine leprosy, a chronic, granulomatous disease similar to human leprosy. Due to the similar clinical manifestations of human and murine leprosy and the difficulty of growing both bacilli axenically, Mycobacterium leprae and M. lepraemurium were once thought to be closely related, although it was later suggested that M. lepraemurium might be related to Mycobacterium avium In this study, the complete genome of M. lepraemurium was sequenced using a combination of PacBio and Illumina sequencing. Phylogenomic analyses confirmed that M. lepraemurium is a distinct species within the M. avium complex (MAC). The M. lepraemurium genome is 4.05 Mb in length, which is considerably smaller than other MAC genomes, and it comprises 2,682 functional genes and 1,139 pseudogenes, which indicates that M. lepraemurium has undergone genome reduction. An error-prone repair homologue of the DNA polymerase III α-subunit was found to be nonfunctional in M. lepraemurium, which might contribute to pseudogene formation due to the accumulation of mutations in nonessential genes. M. lepraemurium has retained the functionality of several genes thought to influence virulence among members of the MAC.IMPORTANCEMycobacterium lepraemurium seems to be evolving toward a minimal set of genes required for an obligatory intracellular lifestyle within its host, a niche seldom adopted by most mycobacteria, as they are free-living. M. lepraemurium could be used as a model to elucidate functions of genes shared with other members of the MAC. Its reduced gene set can be exploited for studying the essentiality of genes in related pathogenic species, which might lead to discovery of common virulence factors or clarify host-pathogen interactions. M. lepraemurium can be cultivated in vitro only under specific conditions and even then with difficulty. Elucidating the metabolic (in)capabilities of M. lepraemurium will help develop suitable axenic media and facilitate genetic studies.
Keywords: Mycobacterium lepraemurium; comparative genomics; genome sequencing; murine leprosy.
Copyright © 2017 Benjak et al.
Figures

Similar articles
-
Highly Reduced Genome of the New Species Mycobacterium uberis, the Causative Agent of Nodular Thelitis and Tuberculoid Scrotitis in Livestock and a Close Relative of the Leprosy Bacilli.mSphere. 2018 Oct 3;3(5):e00405-18. doi: 10.1128/mSphere.00405-18. mSphere. 2018. PMID: 30282756 Free PMC article.
-
Insight into the evolution and origin of leprosy bacilli from the genome sequence of Mycobacterium lepromatosis.Proc Natl Acad Sci U S A. 2015 Apr 7;112(14):4459-64. doi: 10.1073/pnas.1421504112. Epub 2015 Mar 23. Proc Natl Acad Sci U S A. 2015. PMID: 25831531 Free PMC article.
-
Determination of the etiology of presumptive feline leprosy by 16S rRNA gene analysis.J Clin Microbiol. 1997 Oct;35(10):2464-71. doi: 10.1128/jcm.35.10.2464-2471.1997. J Clin Microbiol. 1997. PMID: 9316890 Free PMC article.
-
Mycobacterium leprae and Mycobacterium lepraemurium infections in domestic and wild animals.Rev Sci Tech. 2001 Apr;20(1):219-51. doi: 10.20506/rst.20.1.1271. Rev Sci Tech. 2001. PMID: 11288514 Review.
-
Mycobacterium leprae's evolution and environmental adaptation.Acta Trop. 2019 Sep;197:105041. doi: 10.1016/j.actatropica.2019.105041. Epub 2019 May 30. Acta Trop. 2019. PMID: 31152726 Review.
Cited by
-
Reservoirs and transmission routes of leprosy; A systematic review.PLoS Negl Trop Dis. 2020 Apr 27;14(4):e0008276. doi: 10.1371/journal.pntd.0008276. eCollection 2020 Apr. PLoS Negl Trop Dis. 2020. PMID: 32339201 Free PMC article.
-
Comparative Genomic Analysis of a Methylorubrum rhodesianum MB200 Isolated from Biogas Digesters Provided New Insights into the Carbon Metabolism of Methylotrophic Bacteria.Int J Mol Sci. 2023 Apr 19;24(8):7521. doi: 10.3390/ijms24087521. Int J Mol Sci. 2023. PMID: 37108681 Free PMC article.
-
CAPRIB: a user-friendly tool to study amino acid changes and selection for the exploration of intra-genus evolution.BMC Genomics. 2020 Nov 26;21(1):832. doi: 10.1186/s12864-020-07232-3. BMC Genomics. 2020. PMID: 33243176 Free PMC article.
-
Construction and Analysis of the Complete Genome Sequence of Leprosy Agent Mycobacterium lepromatosis.Microbiol Spectr. 2022 Jun 29;10(3):e0169221. doi: 10.1128/spectrum.01692-21. Epub 2022 Apr 25. Microbiol Spectr. 2022. PMID: 35467405 Free PMC article.
-
Investigating in vivo Mycobacterium avium subsp. paratuberculosis microevolution and mixed strain infections.Microbiol Spectr. 2023 Aug 16;11(5):e0171623. doi: 10.1128/spectrum.01716-23. Online ahead of print. Microbiol Spectr. 2023. PMID: 37584606 Free PMC article.
References
-
- Rojas-Espinosa O. 2009. Murine leprosy revisited, p 97–140. In Tomioka H (ed), Current topics on the profiles of host immunological response to mycobacterial infections, 1st ed. Research Signpost, Trivandrum, Kerala, India.
-
- Stefansky WK. 1902. Zabolevanija u krys, vyzvannyja kislotoupornoj palotsjkoj. Rus Vratsj 47:1726–1727.
-
- Marchoux E, Sorel F. 1912. Recherches sur la lèpre: la lèpre des rats (Lepra murium). Ann Inst Pasteur 56:778–801.
-
- Dean G. 1903. A disease of the rat caused by an acid-fast bacillus. Cent F Bakteriol 34:222–224.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
