

The Molecular Physiopathogenesis of Islet Amyloidosis
- PMID: 29043504
- PMCID: PMC5906204
- DOI: 10.1007/164_2017_62
The Molecular Physiopathogenesis of Islet Amyloidosis
Abstract
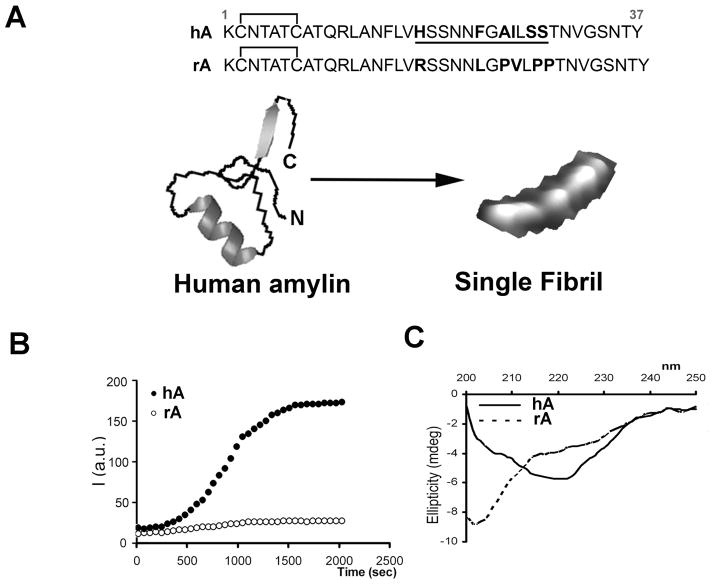
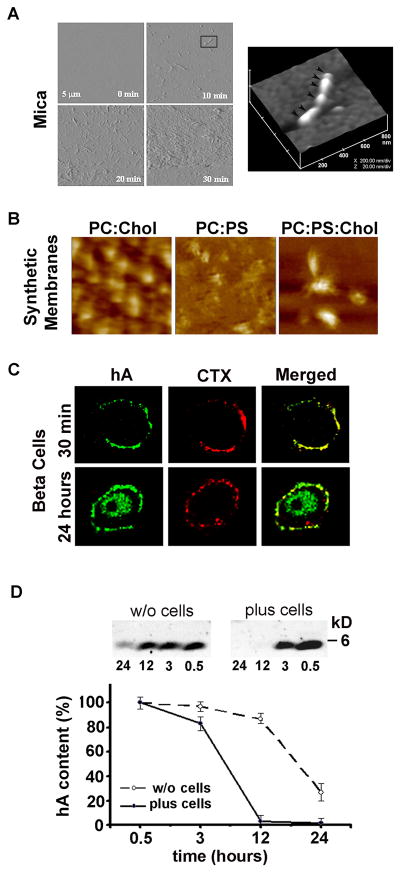
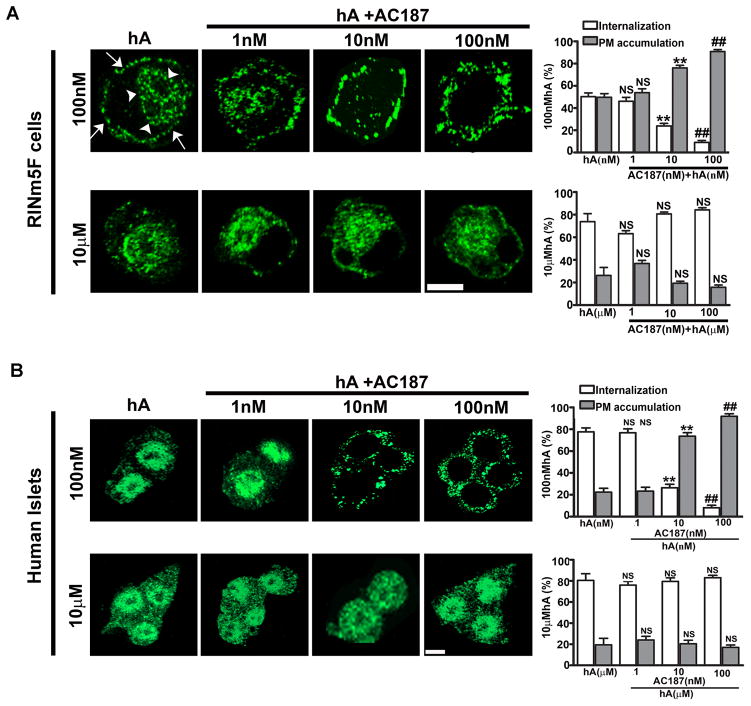
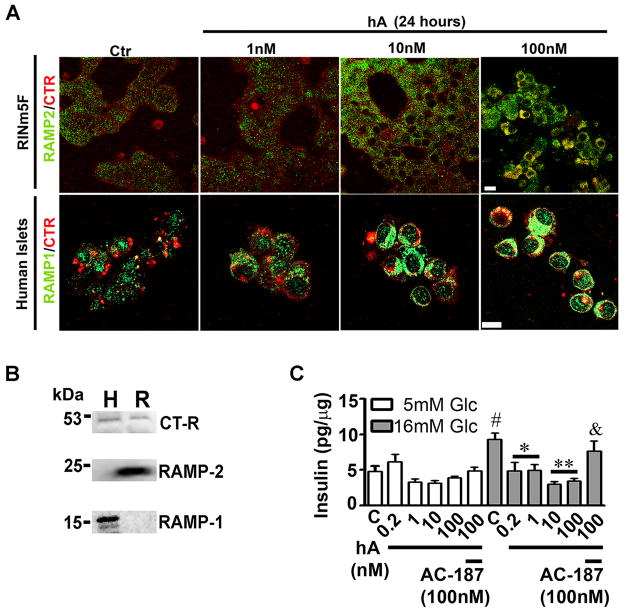
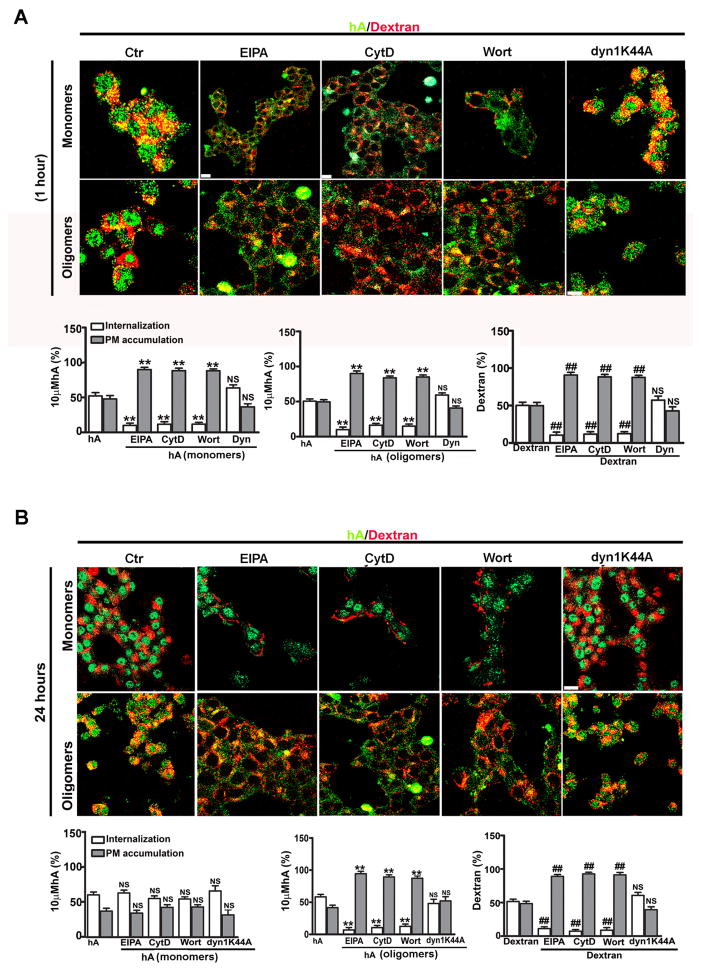
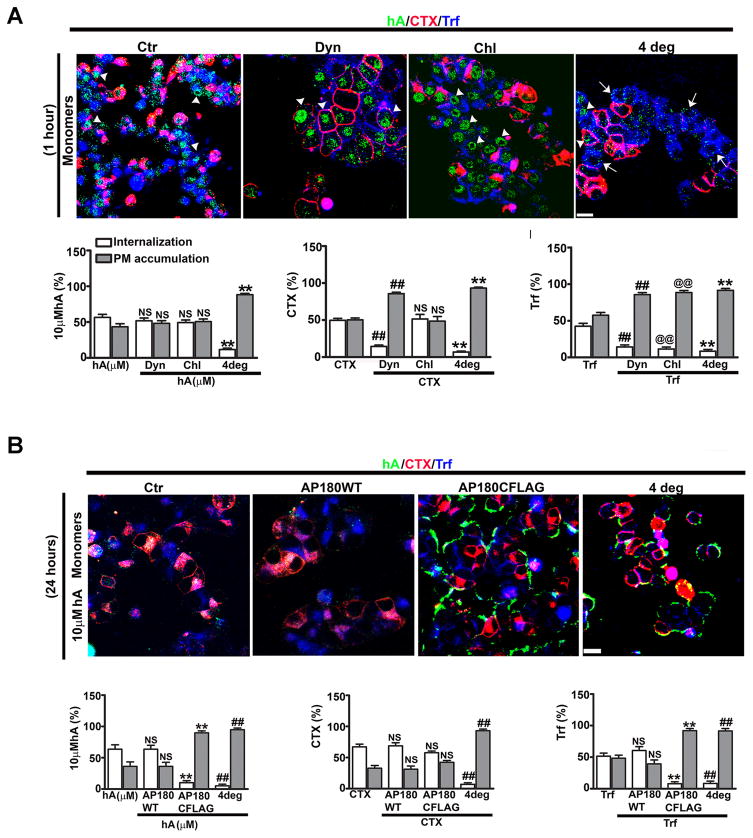
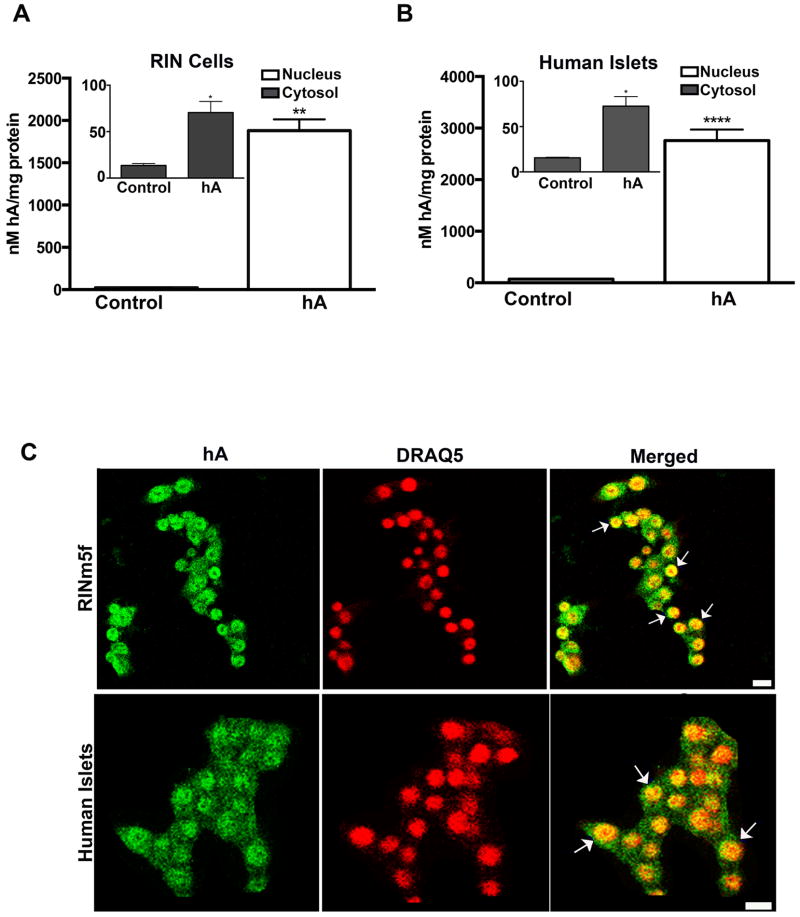
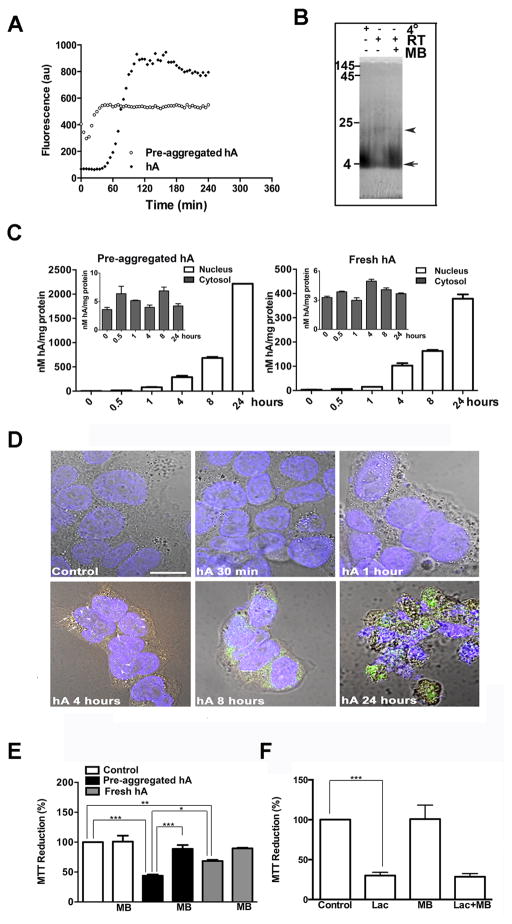
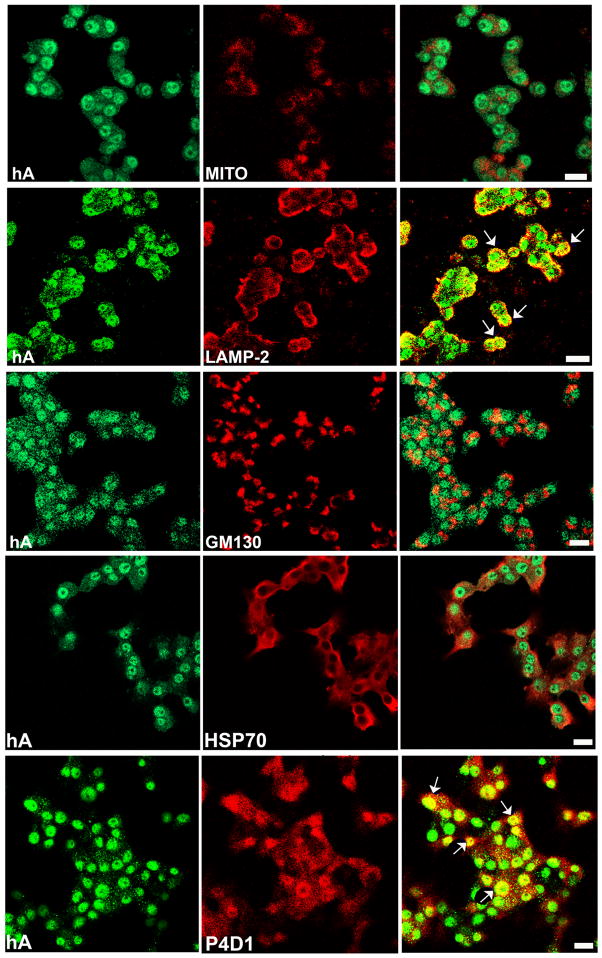
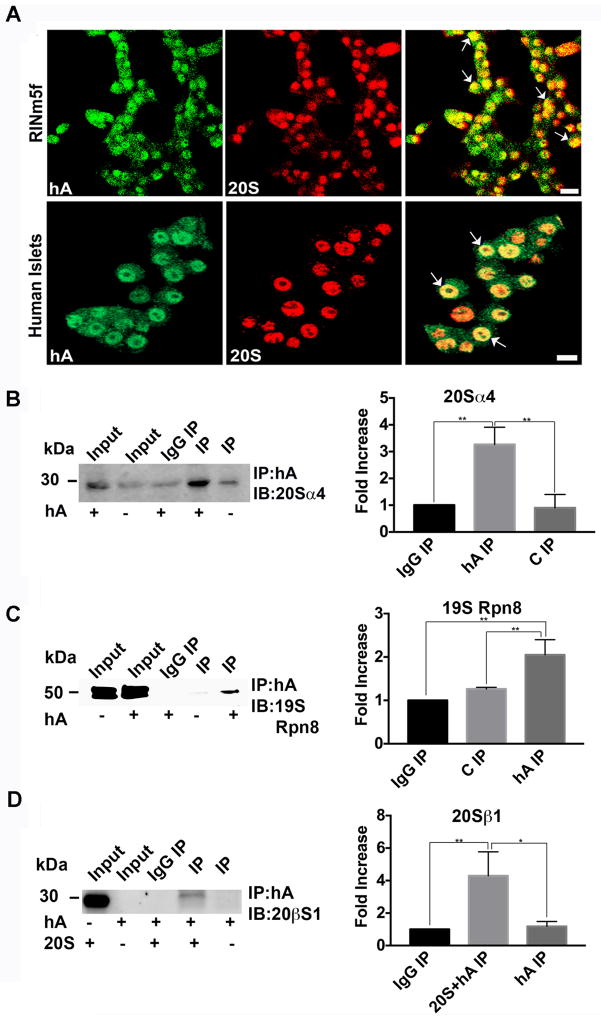
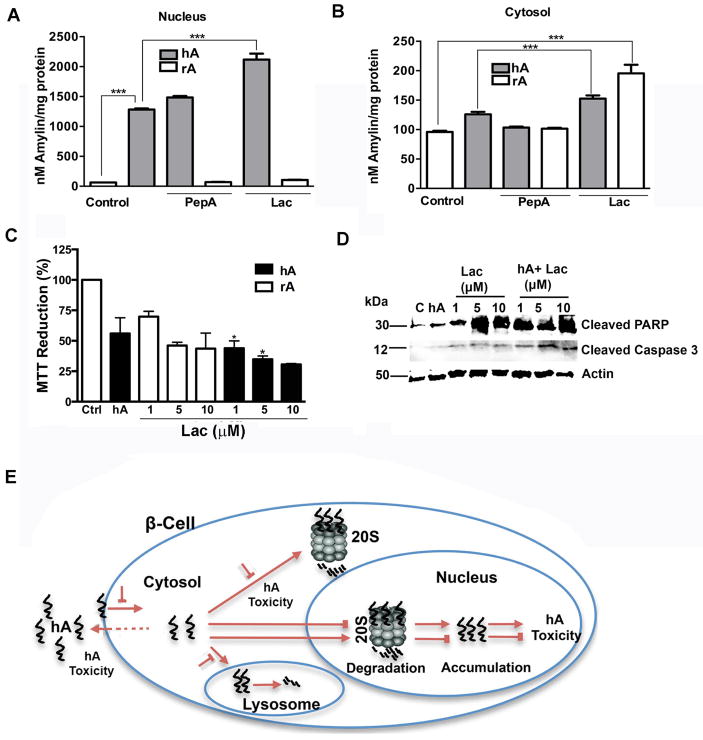
Human islet amyloid polypeptide or amylin (hA) is a 37-amino acid peptide hormone produced and co-secreted with insulin by pancreatic β-cells. Under physiological conditions, hA regulates a broad range of biological processes including insulin release and slowing of gastric emptying, thereby maintaining glucose homeostasis. However, under the pathological conditions associated with type 2 diabetes mellitus (T2DM), hA undergoes a conformational transition from soluble random coil monomers to alpha-helical oligomers and insoluble β-sheet amyloid fibrils or amyloid plaques. There is a positive correlation between hA oligomerization/aggregation, hA toxicity, and diabetes progression. Because the homeostatic balance between hA synthesis, release, and uptake is lost in diabetics and hA aggregation is a hallmark of T2DM, this chapter focuses on the biophysical and cell biology studies investigating molecular mechanisms of hA uptake, trafficking, and degradation in pancreatic cells and its relevance to h's toxicity. We will also discuss the regulatory role of endocytosis and proteolytic pathways in clearance of toxic hA species. Finally, we will discuss potential pharmacological approaches for specific targeting of hA trafficking pathways and toxicity in islet β-cells as potential new avenues toward treatments of T2DM patients.
Keywords: Aggregation; Endocytosis; Human amylin; Islet amyloidosis; Proteasome; Proteotoxicity; Type 2 diabetes mellitus.
Figures
Similar articles
-
Role of Cholesterol and Phospholipids in Amylin Misfolding, Aggregation and Etiology of Islet Amyloidosis.Adv Exp Med Biol. 2015;855:95-116. doi: 10.1007/978-3-319-17344-3_4. Adv Exp Med Biol. 2015. PMID: 26149927 Free PMC article. Review.
-
Islet amyloid polypeptide: a review of its biology and potential roles in the pathogenesis of diabetes mellitus.Vet Pathol. 1993 Jul;30(4):317-32. doi: 10.1177/030098589303000401. Vet Pathol. 1993. PMID: 8212454 Review.
-
Molecular Mechanisms of Amylin Turnover, Misfolding and Toxicity in the Pancreas.Molecules. 2022 Feb 2;27(3):1021. doi: 10.3390/molecules27031021. Molecules. 2022. PMID: 35164285 Free PMC article. Review.
-
Human Amylin: From Pathology to Physiology and Pharmacology.Curr Protein Pept Sci. 2019;20(9):944-957. doi: 10.2174/1389203720666190328111833. Curr Protein Pept Sci. 2019. PMID: 30919775 Review.
-
Islet amyloid polypeptide, islet amyloid, and diabetes mellitus.Physiol Rev. 2011 Jul;91(3):795-826. doi: 10.1152/physrev.00042.2009. Physiol Rev. 2011. PMID: 21742788 Review.
Cited by
-
Novel Hominid-Specific IAPP Isoforms: Potential Biomarkers of Early Alzheimer's Disease and Inhibitors of Amyloid Formation.Biomolecules. 2023 Jan 13;13(1):167. doi: 10.3390/biom13010167. Biomolecules. 2023. PMID: 36671553 Free PMC article.
-
Organisation of the human pancreas in health and in diabetes.Diabetologia. 2020 Oct;63(10):1966-1973. doi: 10.1007/s00125-020-05203-7. Epub 2020 Sep 7. Diabetologia. 2020. PMID: 32894306 Free PMC article. Review.
-
Type 2 Diabetes Mellitus: Pathogenic Features and Experimental Models in Rodents.Acta Naturae. 2022 Jul-Sep;14(3):57-68. doi: 10.32607/actanaturae.11751. Acta Naturae. 2022. PMID: 36348712 Free PMC article.
References
-
- Ancsin JB. Amyloidogenesis: historical and modern observations point to heparan sulfate proteoglycans as a major culprit. Amyloid. 2003;10(2):67–79. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
