

Modeling and Preventing Progressive Hearing Loss in Usher Syndrome III
- PMID: 29044151
- PMCID: PMC5647385
- DOI: 10.1038/s41598-017-13620-9
Modeling and Preventing Progressive Hearing Loss in Usher Syndrome III
Abstract
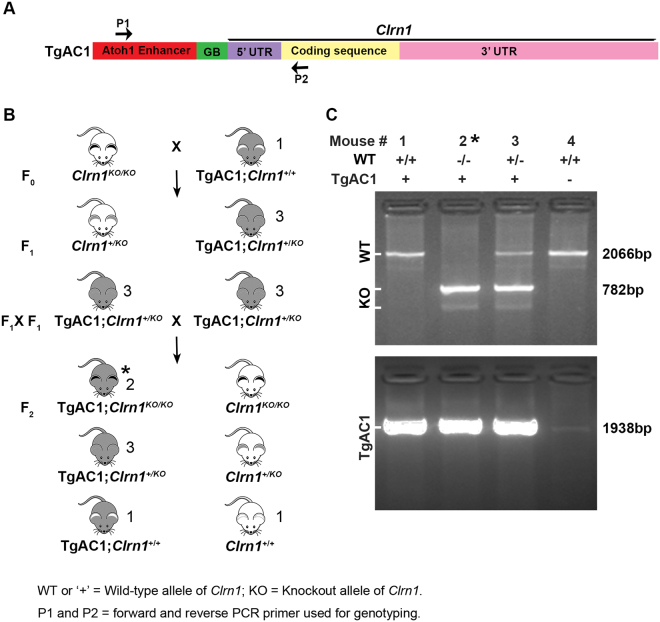
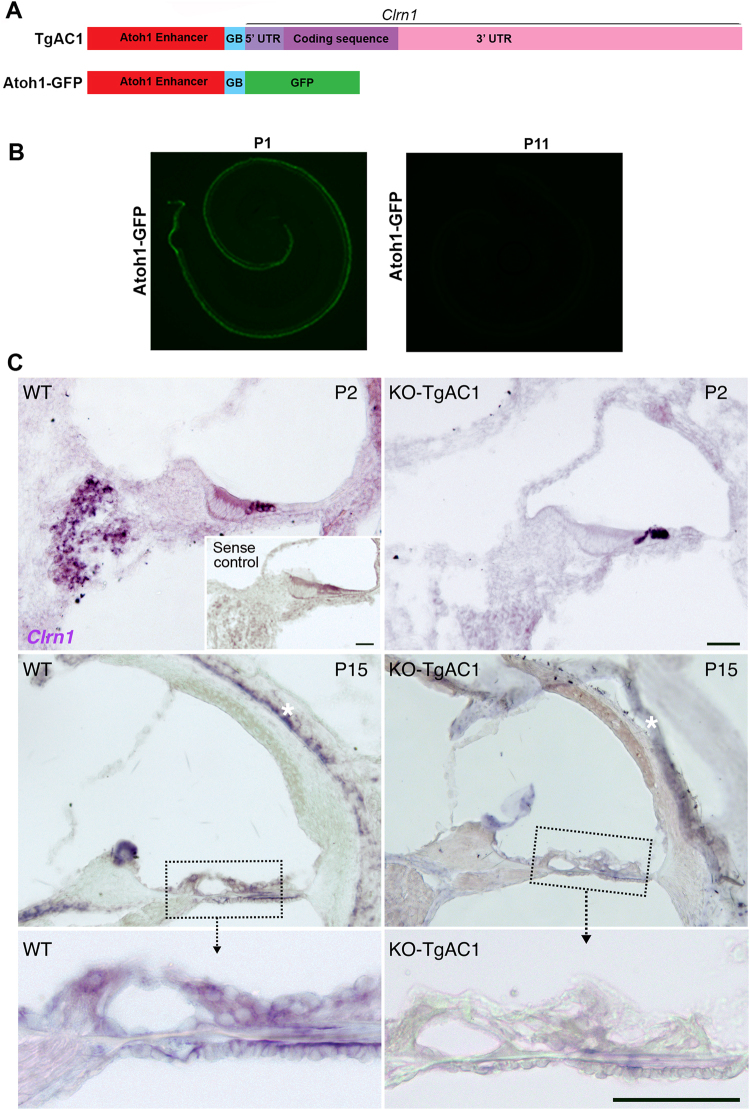
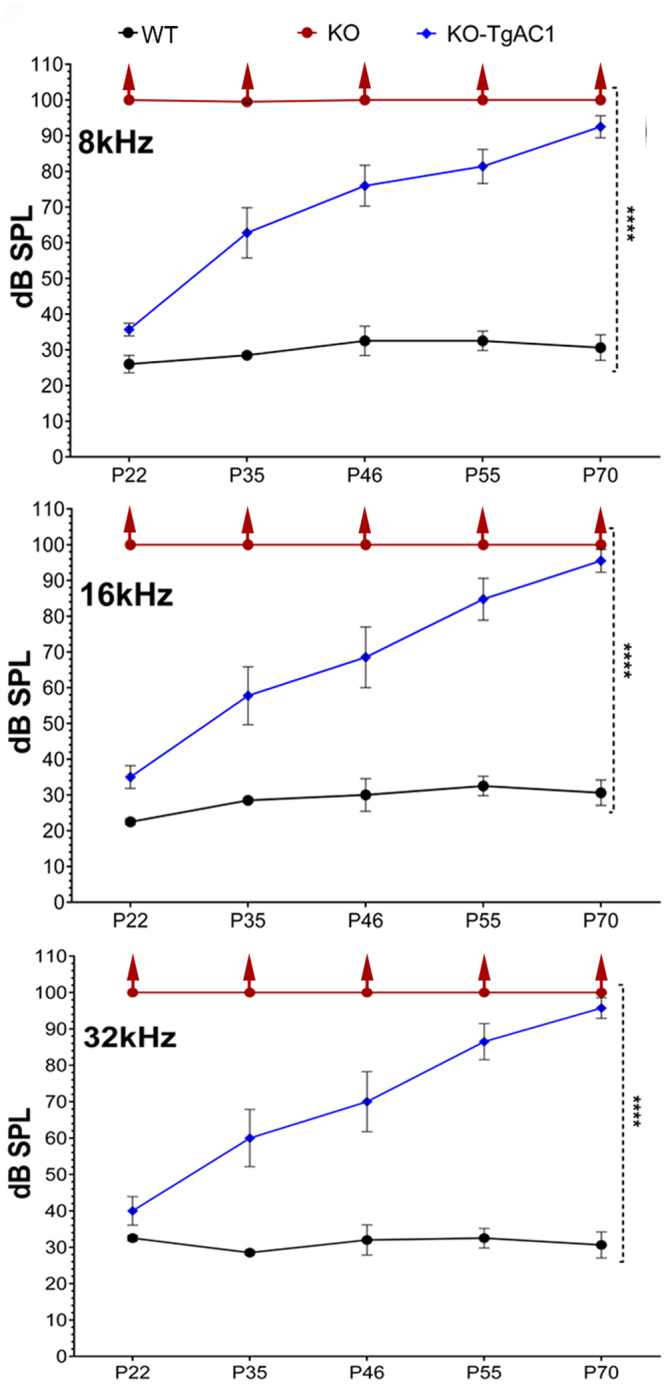
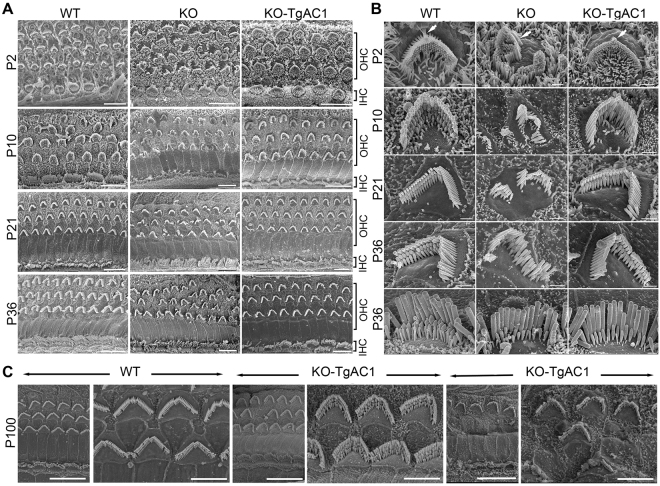
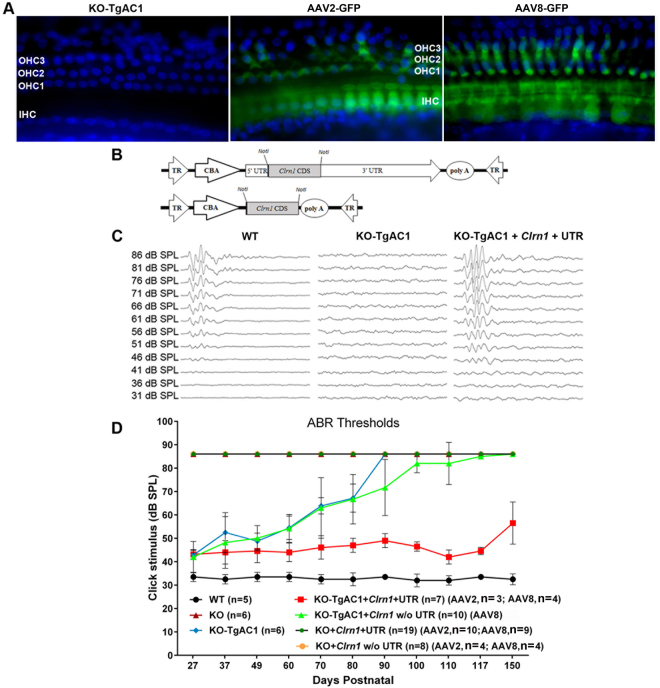
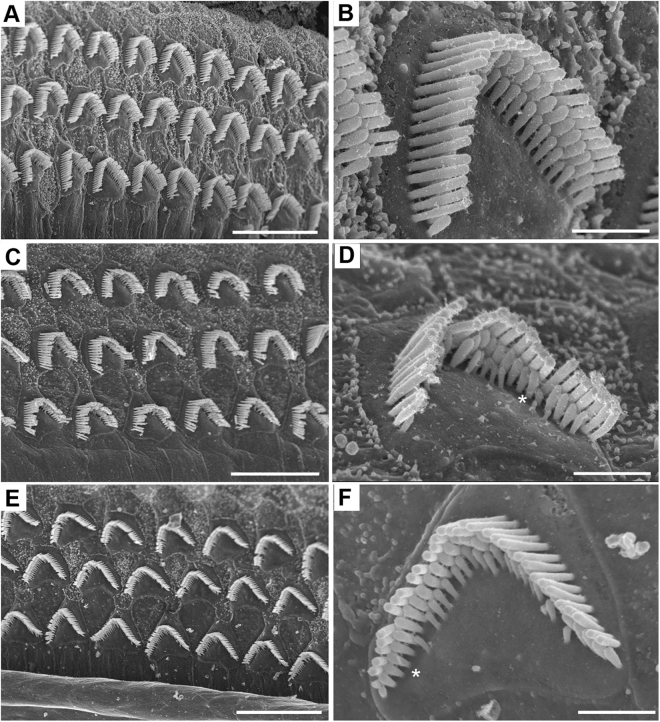
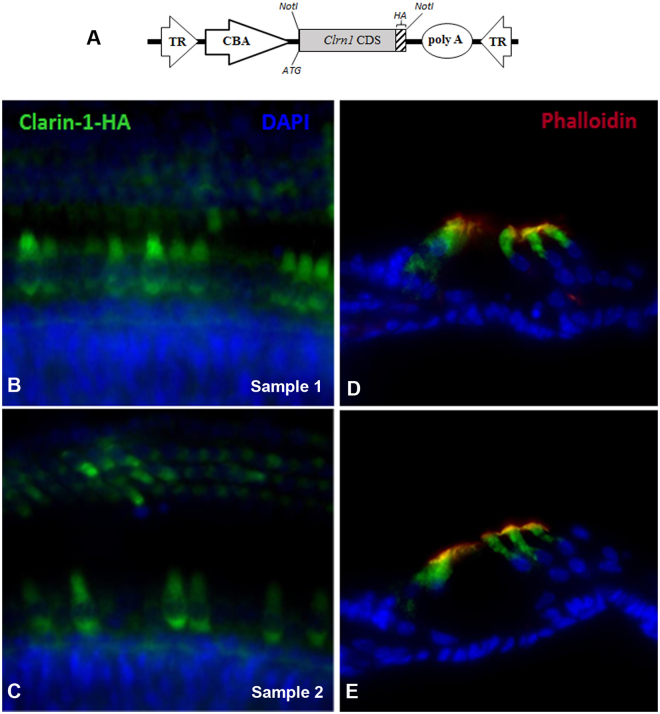
Usher syndrome type III (USH3) characterized by progressive loss of vision and hearing is caused by mutations in the clarin-1 gene (CLRN1). Clrn1 knockout (KO) mice develop hair cell defects by postnatal day 2 (P2) and are deaf by P21-P25. Early onset profound hearing loss in KO mice and lack of information about the cochlear cell type that requires Clrn1 expression pose challenges to therapeutic investigation. We generated KO mice harboring a transgene, TgAC1, consisting of Clrn1-UTR (Clrn1 cDNA including its 5' and 3' UTR) under the control of regulatory elements (Atoh1 3' enhancer/β-globin basal promoter) to direct expression of Clrn1 in hair cells during development and down regulate it postnatally. The KO-TgAC1 mice displayed delayed onset progressive hearing loss associated with deterioration of the hair bundle structure, leading to the hypothesis that hair cell expression of Clrn1 is essential for postnatal preservation of hair cell structure and hearing. Consistent with that hypothesis, perinatal transfection of hair cells in KO-TgAC1 mice with a single injection of AAV-Clrn1-UTR vector showed correlative preservation of the hair bundle structure and hearing through adult life. Further, the efficacy of AAV-Clrn1 vector was significantly attenuated, revealing the potential importance of UTR in gene therapy.
Conflict of interest statement
The authors declare the following competing financial interests: The mouse
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
