

Outside-in HLA class I signaling regulates ICAM-1 clustering and endothelial cell-monocyte interactions via mTOR in transplant antibody-mediated rejection
- PMID: 29045076
- PMCID: PMC5904014
- DOI: 10.1111/ajt.14544
Outside-in HLA class I signaling regulates ICAM-1 clustering and endothelial cell-monocyte interactions via mTOR in transplant antibody-mediated rejection
Abstract
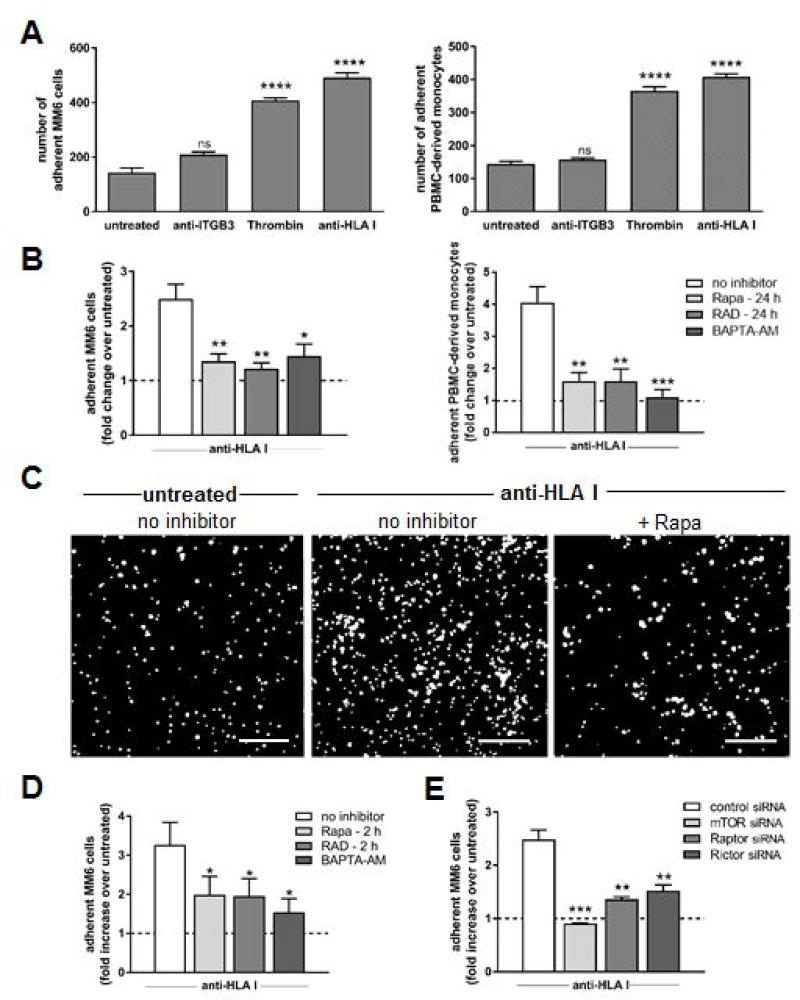
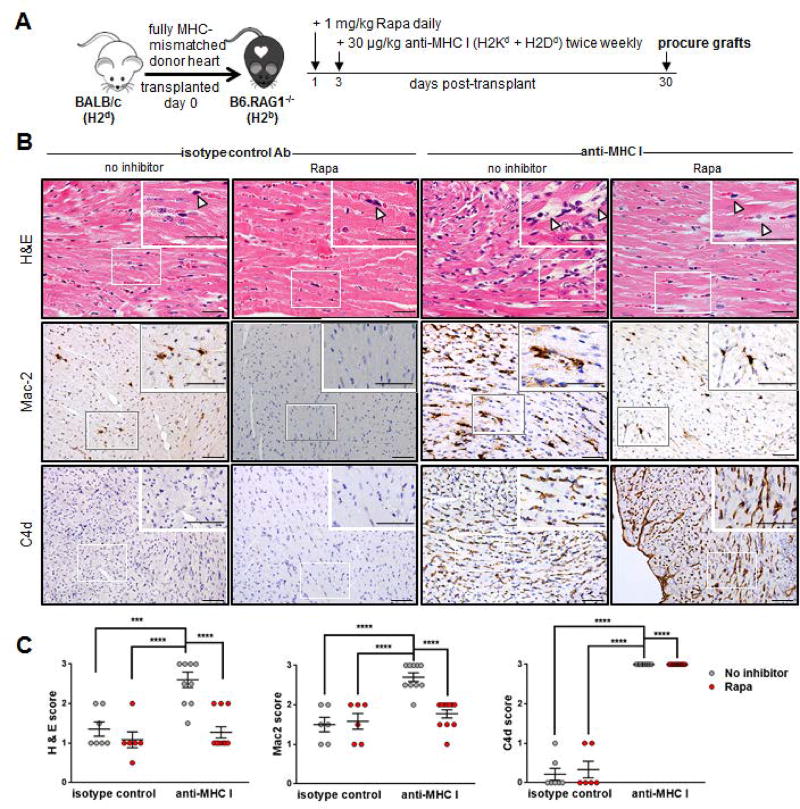
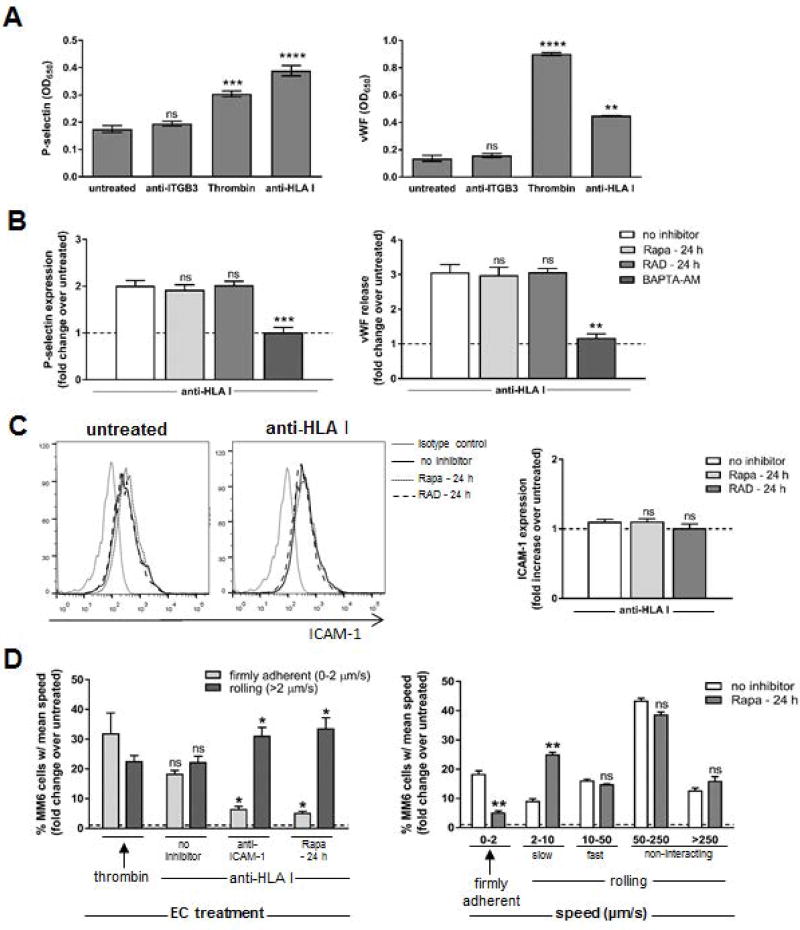
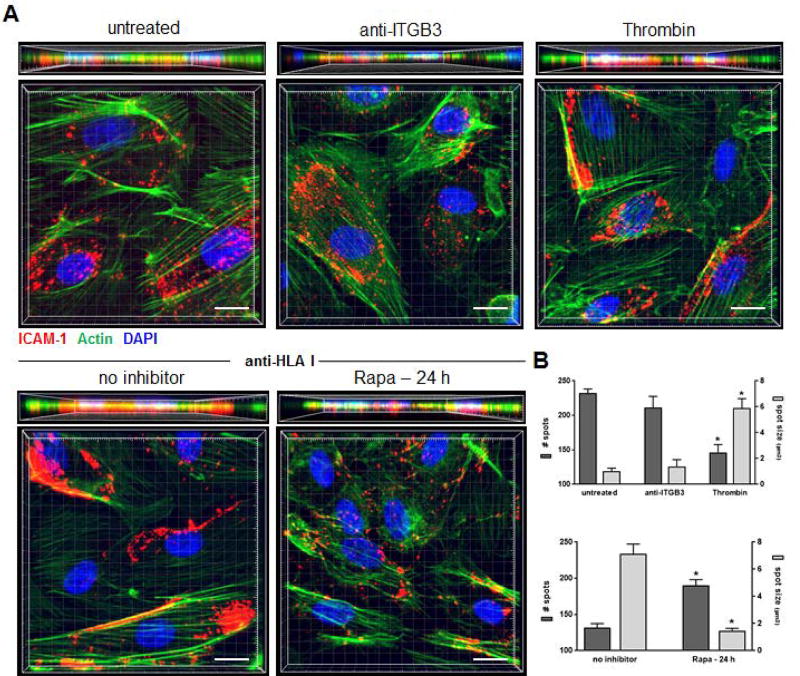
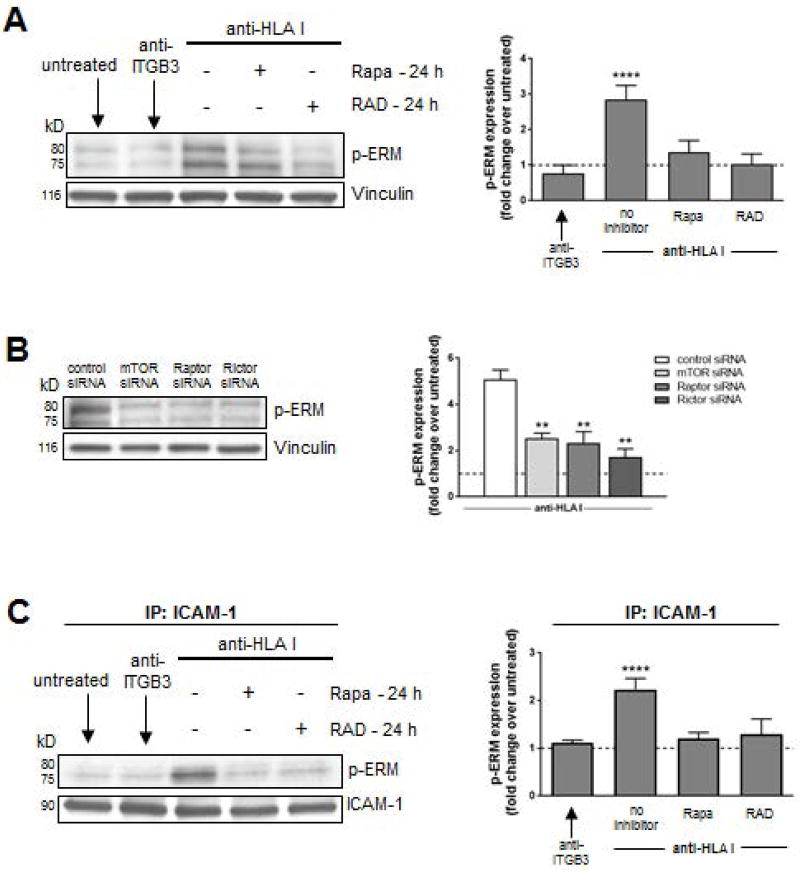
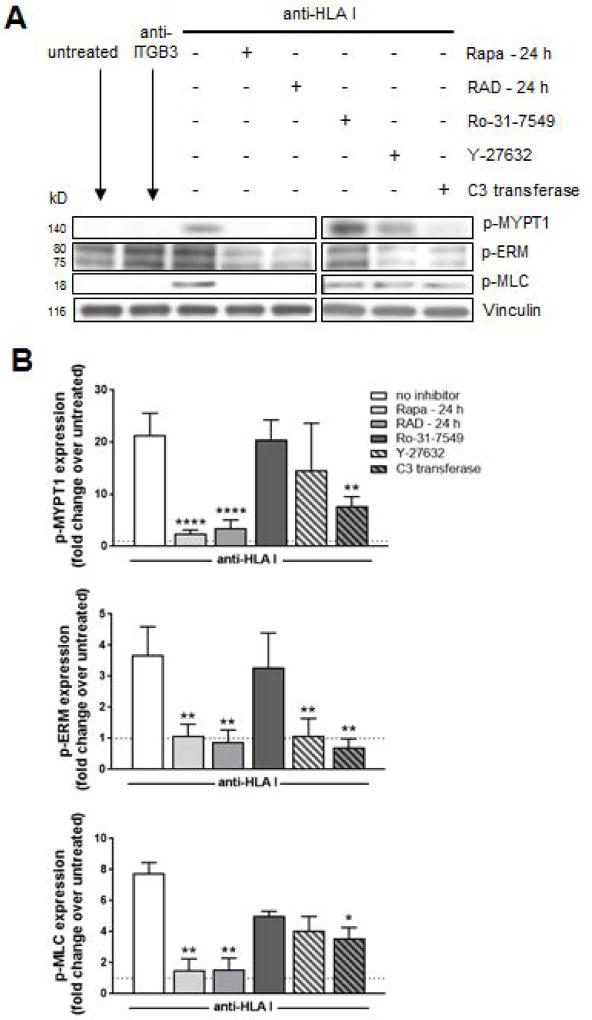
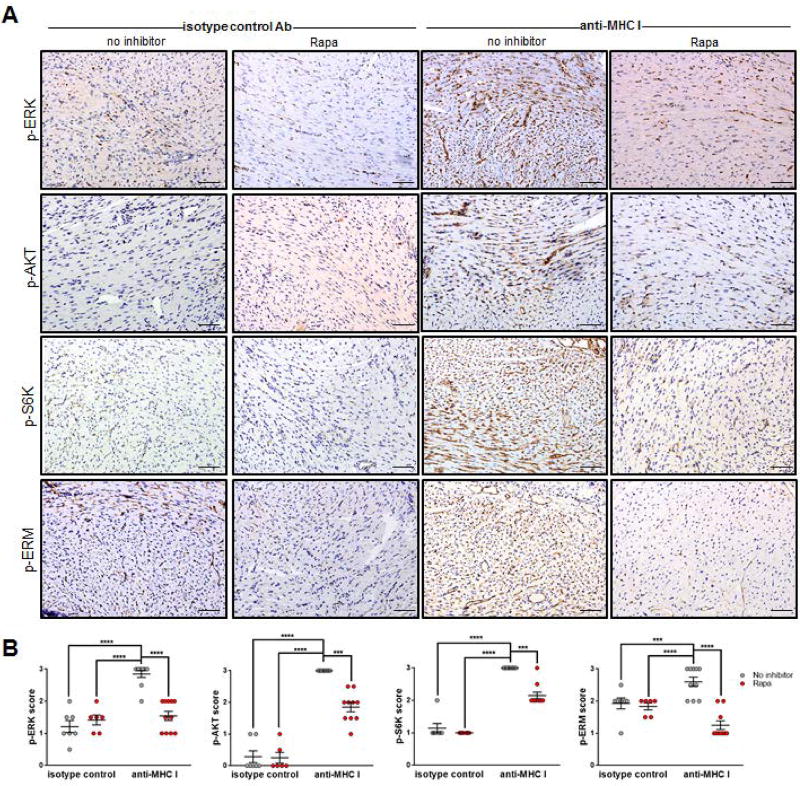
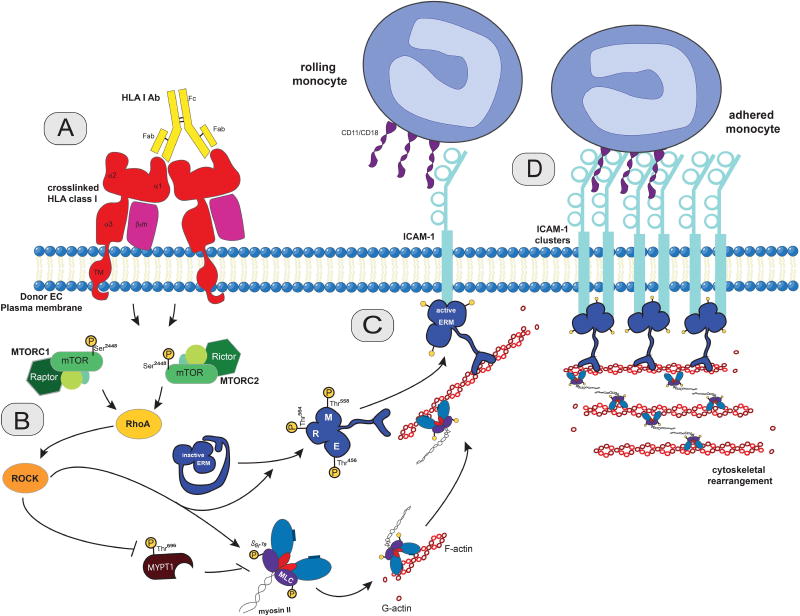
Antibody-mediated rejection (AMR) resulting in transplant allograft vasculopathy (TAV) is the major obstacle for long-term survival of solid organ transplants. AMR is caused by donor-specific antibodies to HLA, which contribute to TAV by initiating outside-in signaling transduction pathways that elicit monocyte recruitment to activated endothelium. Mechanistic target of rapamycin (mTOR) inhibitors can attenuate TAV; therefore, we sought to understand the mechanistic underpinnings of mTOR signaling in HLA class I Ab-mediated endothelial cell activation and monocyte recruitment. We used an in vitro model to assess monocyte binding to HLA I Ab-activated endothelial cells and found mTOR inhibition reduced ezrin/radixin/moesin (ERM) phosphorylation, intercellular adhesion molecule 1 (ICAM-1) clustering, and monocyte firm adhesion to HLA I Ab-activated endothelium. Further, in a mouse model of AMR, in which C57BL/6. RAG1-/- recipients of BALB/c cardiac allografts were passively transferred with donor-specific MHC I antibodies, mTOR inhibition significantly reduced vascular injury, ERM phosphorylation, and macrophage infiltration of the allograft. Taken together, these studies indicate mTOR inhibition suppresses ERM phosphorylation in endothelial cells, which impedes ICAM-1 clustering in response to HLA class I Ab and prevents macrophage infiltration into cardiac allografts. These findings indicate a novel therapeutic application for mTOR inhibitors to disrupt endothelial cell-monocyte interactions during AMR.
Keywords: alloantibody; animal models: murine; basic (laboratory) research/science; cellular biology; immunosuppressant - mechanistic target of rapamycin (mTOR); immunosuppression/immune modulation; macrophage/monocyte biology; organ transplantation in general; translational research/science; vasculopathy.
© 2017 The American Society of Transplantation and the American Society of Transplant Surgeons.
Conflict of interest statement
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. EFR received funding for this research through the Pfizer Arts grant. The other authors have no conflicts of interest to disclose.
Figures
Similar articles
-
HLA class I antibodies trigger increased adherence of monocytes to endothelial cells by eliciting an increase in endothelial P-selectin and, depending on subclass, by engaging FcγRs.J Immunol. 2013 Jun 15;190(12):6635-50. doi: 10.4049/jimmunol.1201434. Epub 2013 May 20. J Immunol. 2013. PMID: 23690477 Free PMC article.
-
Antibody-induced vascular inflammation skews infiltrating macrophages to a novel remodeling phenotype in a model of transplant rejection.Am J Transplant. 2020 Oct;20(10):2686-2702. doi: 10.1111/ajt.15934. Epub 2020 May 22. Am J Transplant. 2020. PMID: 32320528 Free PMC article.
-
Blockade of p-selectin is sufficient to reduce MHC I antibody-elicited monocyte recruitment in vitro and in vivo.Am J Transplant. 2013 Feb;13(2):299-311. doi: 10.1111/ajt.12016. Epub 2012 Dec 27. Am J Transplant. 2013. PMID: 23279566 Free PMC article.
-
Interplay between immune responses to HLA and non-HLA self-antigens in allograft rejection.Hum Immunol. 2013 Nov;74(11):1478-85. doi: 10.1016/j.humimm.2013.07.002. Epub 2013 Jul 19. Hum Immunol. 2013. PMID: 23876679 Free PMC article. Review.
-
mTOR inhibitors and risk of chronic antibody-mediated rejection after kidney transplantation: where are we now?Transpl Int. 2017 Jul;30(7):647-657. doi: 10.1111/tri.12975. Transpl Int. 2017. PMID: 28445619 Review.
Cited by
-
An in vitro model of antibody-mediated injury to glomerular endothelial cells: Upregulation of MHC class II and adhesion molecules.Transpl Immunol. 2020 Feb;58:101261. doi: 10.1016/j.trim.2019.101261. Epub 2019 Dec 27. Transpl Immunol. 2020. PMID: 31887408 Free PMC article.
-
Vascular Signaling in Allogenic Solid Organ Transplantation - The Role of Endothelial Cells.Front Physiol. 2020 May 8;11:443. doi: 10.3389/fphys.2020.00443. eCollection 2020. Front Physiol. 2020. PMID: 32457653 Free PMC article. Review.
-
Everolimus plus reduced calcineurin inhibitor prevents de novo anti-HLA antibodies and humoral rejection in kidney transplant recipients: 12-month results from the ATHENA study.Front Transplant. 2023 Oct 27;2:1264903. doi: 10.3389/frtra.2023.1264903. eCollection 2023. Front Transplant. 2023. PMID: 38993866 Free PMC article.
-
Classic and Current Opinions in Human Organ and Tissue Transplantation.Cureus. 2022 Nov 1;14(11):e30982. doi: 10.7759/cureus.30982. eCollection 2022 Nov. Cureus. 2022. PMID: 36337306 Free PMC article. Review.
-
Protection of transplants against antibody-mediated injuries: from xenotransplantation to allogeneic transplantation, mechanisms and therapeutic insights.Front Immunol. 2022 Aug 5;13:932242. doi: 10.3389/fimmu.2022.932242. eCollection 2022. Front Immunol. 2022. PMID: 35990687 Free PMC article. Review.
References
-
- Colvin RB. Antibody-mediated renal allograft rejection: Diagnosis and pathogenesis. Journal of the American Society of Nephrology. 2007;18(4):1046–1056. - PubMed
-
- Berry GJ, Burke MM, Andersen C, Bruneval P, Fedrigo M, Fishbein MC, et al. The 2013 International Society for Heart and Lung Transplantation Working Formulation for the standardization of nomenclature in the pathologic diagnosis of antibody-mediated rejection in heart transplantation. Journal of Heart and Lung Transplantation. 2013;32(12):1147–1162. - PubMed
-
- Abrahimi P, Liu R, Pober JS. Blood Vessels in Allotransplantation. American Journal of Transplantation. 2015;15(7):1748–1754. - PubMed
-
- Fishbein GA, Fishbein MC. Morphologic and immunohistochemical findings in antibody-mediated rejection of the cardiac allograft. Human Immunology. 2012;73(12):1213–1217. - PubMed
-
- Loupy A, Toquet C, Rouvier P, Beuscart T, Bories MC, Varnous S, et al. Late Failing Heart Allografts: Pathology of Cardiac Allograft Vasculopathy and Association With Antibody-Mediated Rejection. American Journal of Transplantation. 2016;16(1):111–120. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
