

Inflammatory memory sensitizes skin epithelial stem cells to tissue damage
- PMID: 29045388
- PMCID: PMC5808576
- DOI: 10.1038/nature24271
Inflammatory memory sensitizes skin epithelial stem cells to tissue damage
Erratum in
-
Author Correction: Inflammatory memory sensitizes skin epithelial stem cells to tissue damage.Nature. 2018 Aug;560(7716):E2. doi: 10.1038/s41586-018-0229-5. Nature. 2018. PMID: 29973713
Abstract
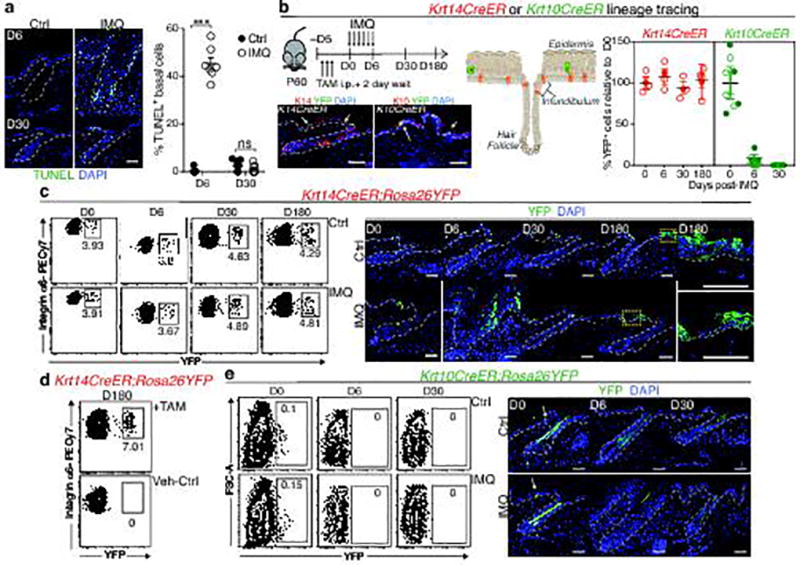
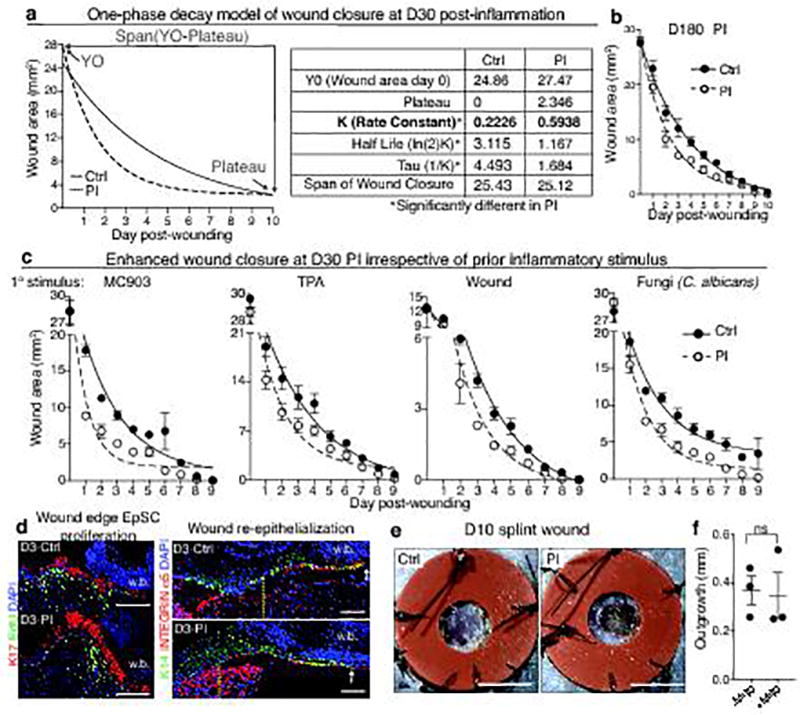
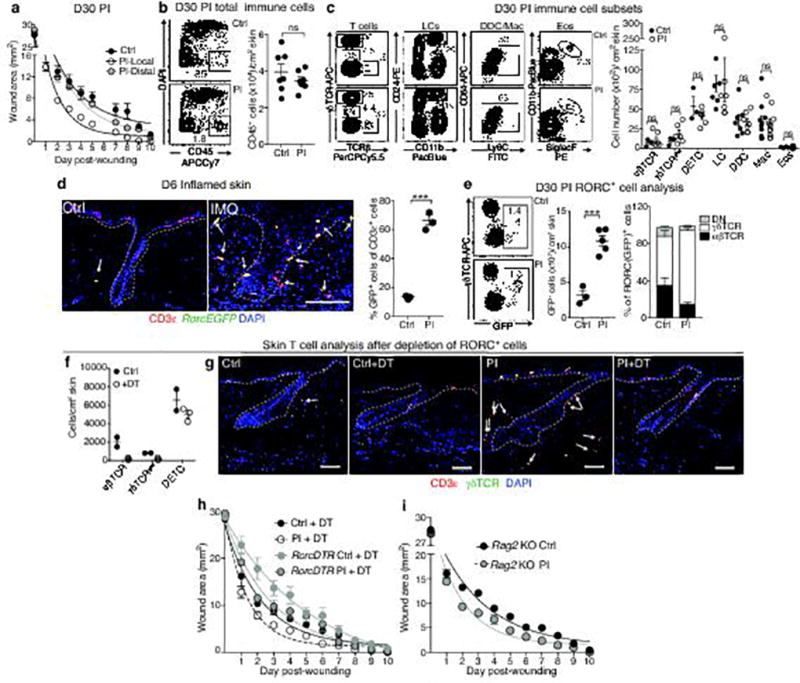
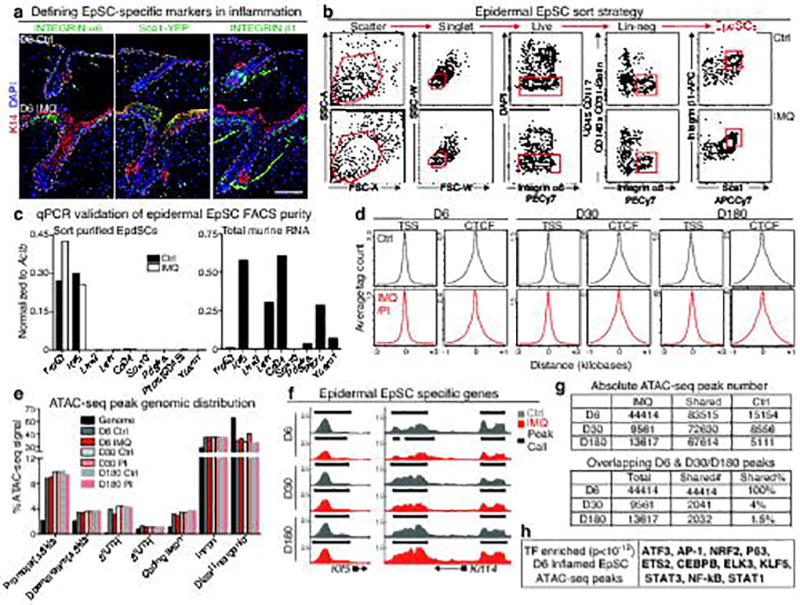
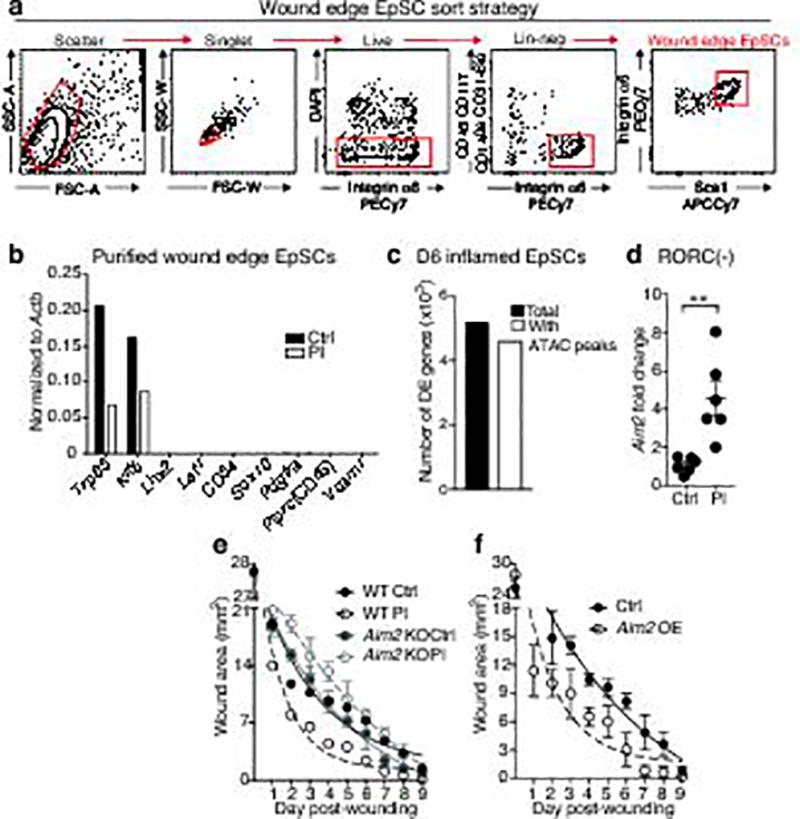
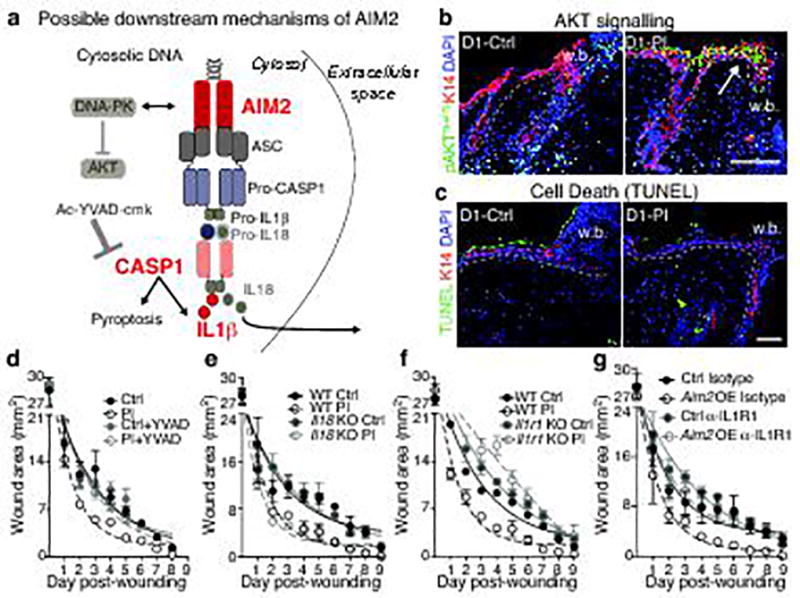
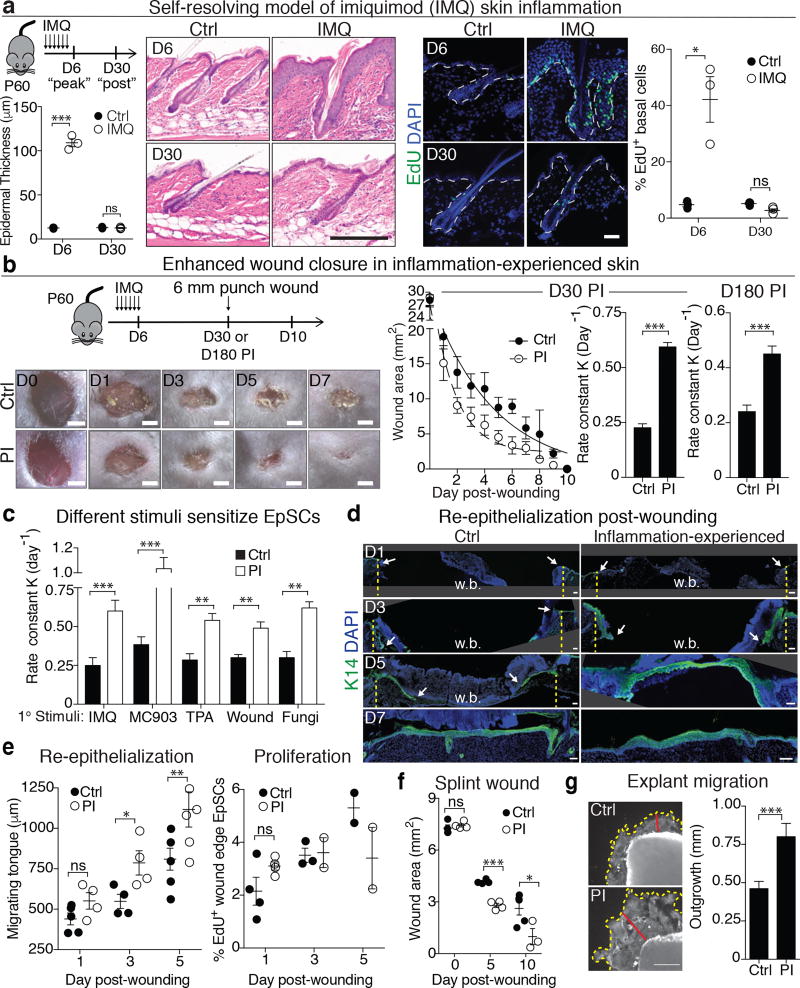
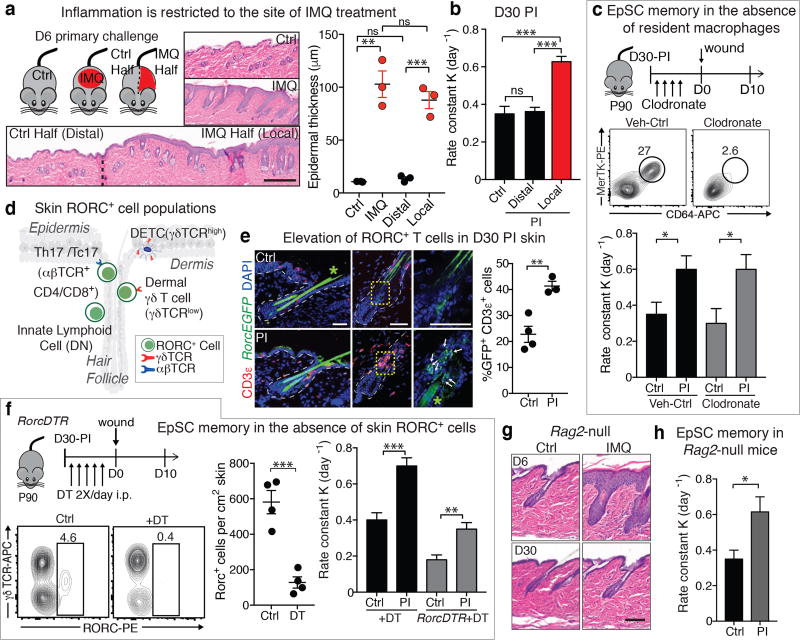
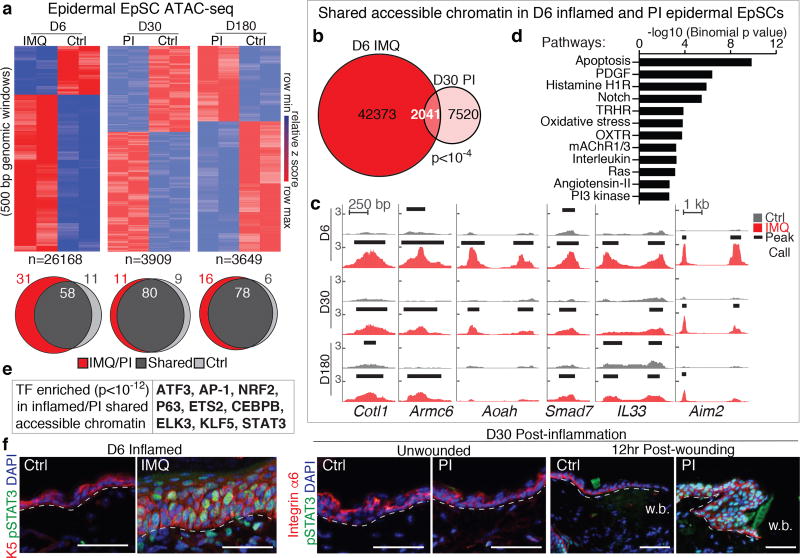
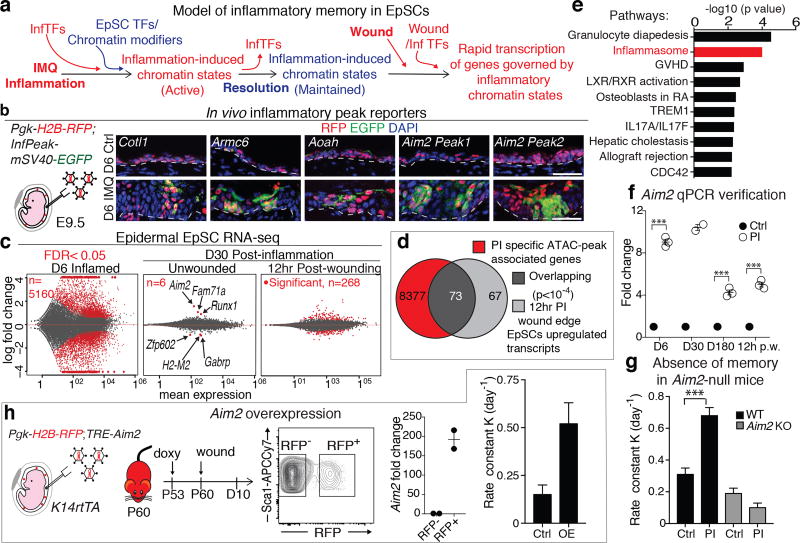
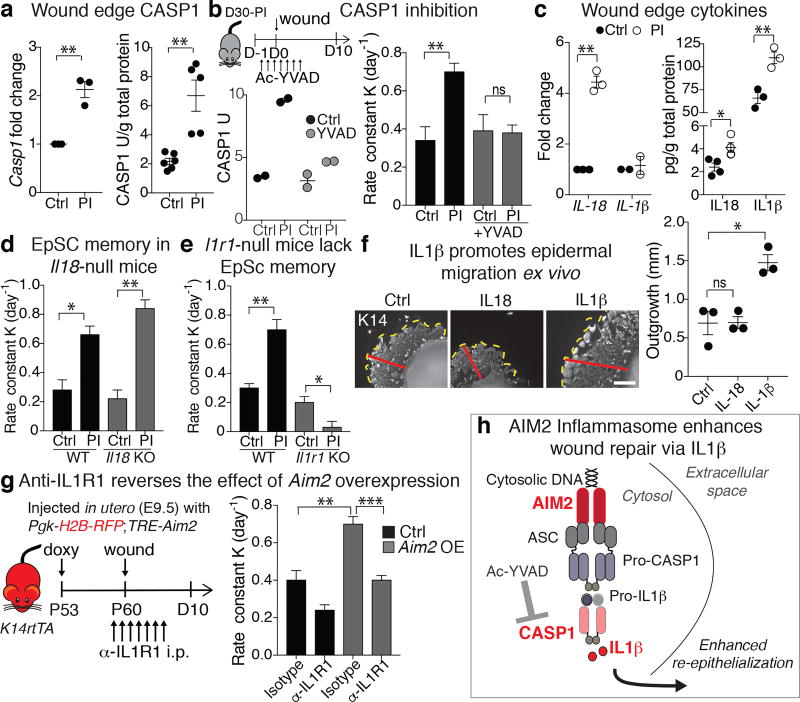
The skin barrier is the body's first line of defence against environmental assaults, and is maintained by epithelial stem cells (EpSCs). Despite the vulnerability of EpSCs to inflammatory pressures, neither the primary response to inflammation nor its enduring consequences are well understood. Here we report a prolonged memory to acute inflammation that enables mouse EpSCs to hasten barrier restoration after subsequent tissue damage. This functional adaptation does not require skin-resident macrophages or T cells. Instead, EpSCs maintain chromosomal accessibility at key stress response genes that are activated by the primary stimulus. Upon a secondary challenge, genes governed by these domains are transcribed rapidly. Fuelling this memory is Aim2, which encodes an activator of the inflammasome. The absence of AIM2 or its downstream effectors, caspase-1 and interleukin-1β, erases the ability of EpSCs to recollect inflammation. Although EpSCs benefit from inflammatory tuning by heightening their responsiveness to subsequent stressors, this enhanced sensitivity probably increases their susceptibility to autoimmune and hyperproliferative disorders, including cancer.
Figures
Comment in
-
Inflammation: Memory beyond immunity.Nature. 2017 Oct 26;550(7677):460-461. doi: 10.1038/nature24154. Epub 2017 Oct 18. Nature. 2017. PMID: 29045392 No abstract available.
-
Inflammation: Inflammatory memory is skin deep.Nat Rev Immunol. 2017 Dec;17(12):731. doi: 10.1038/nri.2017.127. Epub 2017 Nov 7. Nat Rev Immunol. 2017. PMID: 29109540 No abstract available.
-
Locked and Loaded: Inflammation Training Prepares Skin Epithelial Stem Cells for Trauma.Cell Stem Cell. 2017 Dec 7;21(6):715-717. doi: 10.1016/j.stem.2017.11.013. Cell Stem Cell. 2017. PMID: 29220664
-
I Remember You: Epigenetic Priming in Epithelial Stem Cells.Immunity. 2017 Dec 19;47(6):1019-1021. doi: 10.1016/j.immuni.2017.12.005. Immunity. 2017. PMID: 29262345
-
Transplanted stem cells survive a long time: do they make you sick?J R Soc Med. 2019 Oct;112(10):412-414. doi: 10.1177/0141076819851657. Epub 2019 Sep 17. J R Soc Med. 2019. PMID: 31526208 Free PMC article. No abstract available.
References
-
- Kanneganti T-D, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
